

A hypomorphic model of CPS1 deficiency for investigating the effects of hyperammonemia on the developing nervous system
- PMID: 40421838
- PMCID: PMC12208401
- DOI: 10.1242/dmm.052303
A hypomorphic model of CPS1 deficiency for investigating the effects of hyperammonemia on the developing nervous system
Abstract
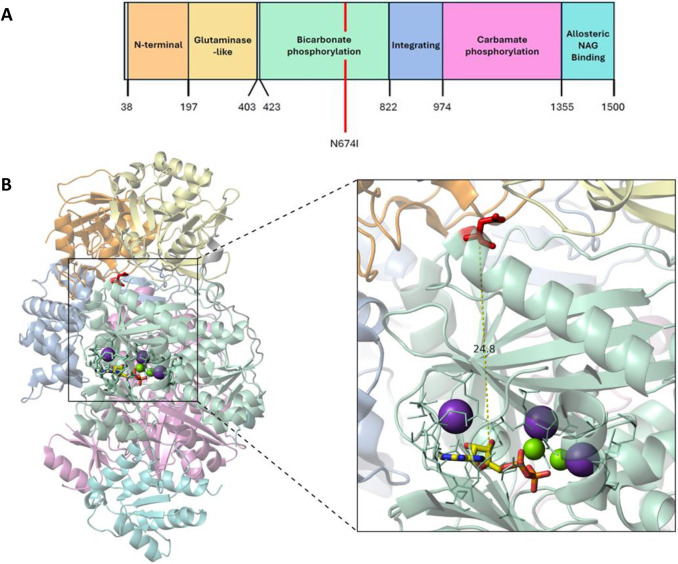
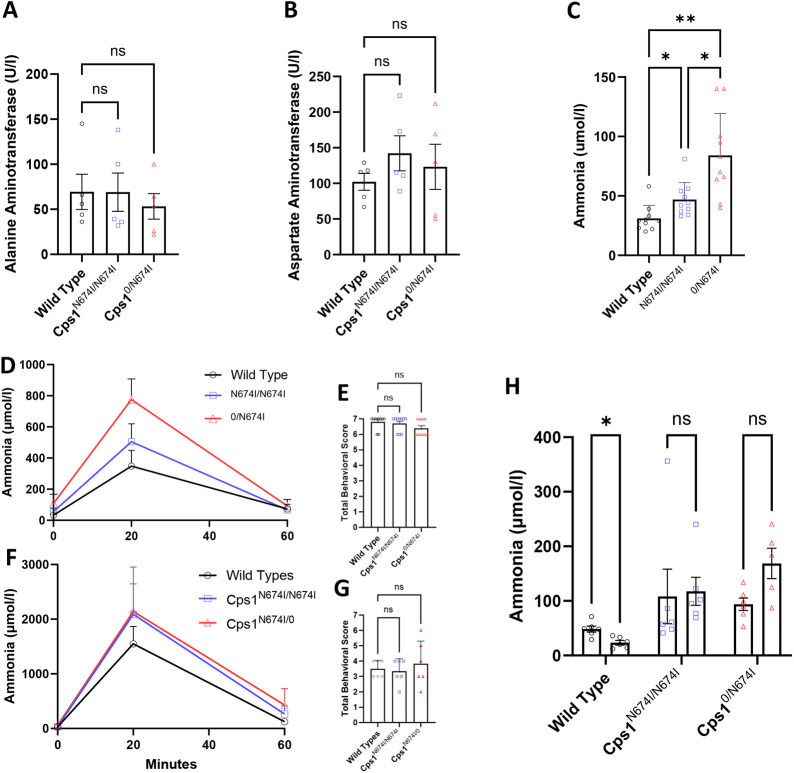
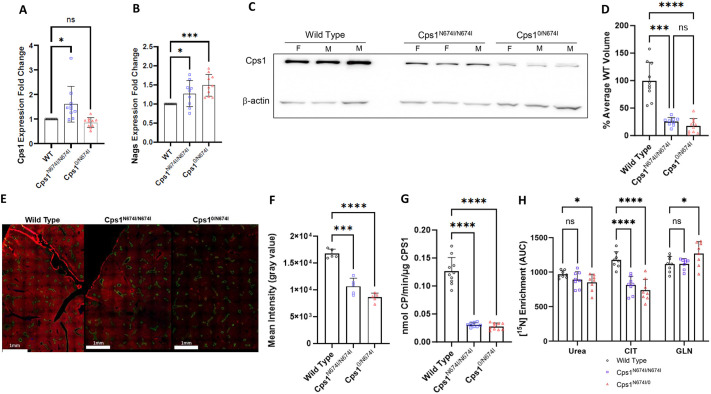
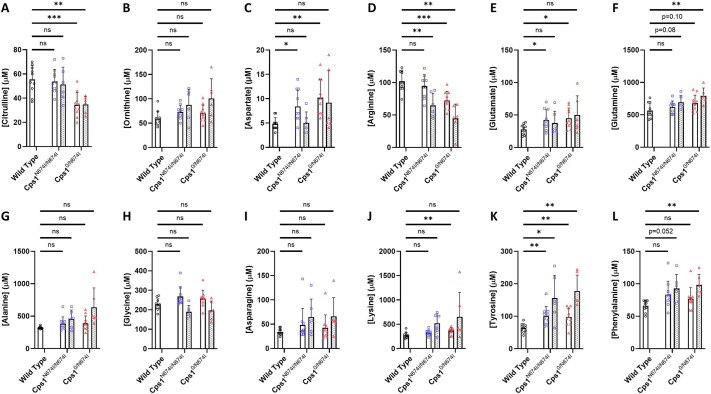
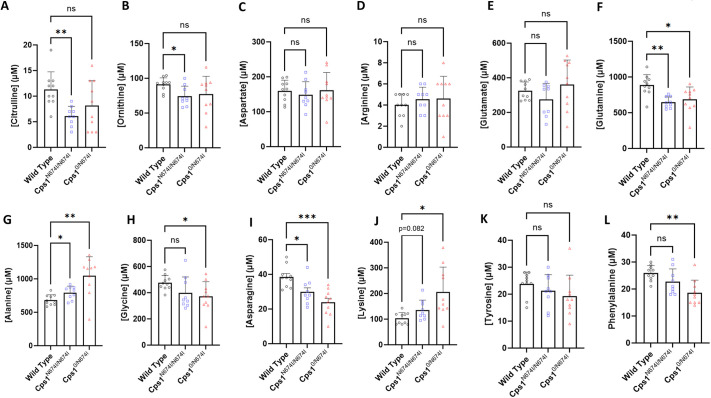
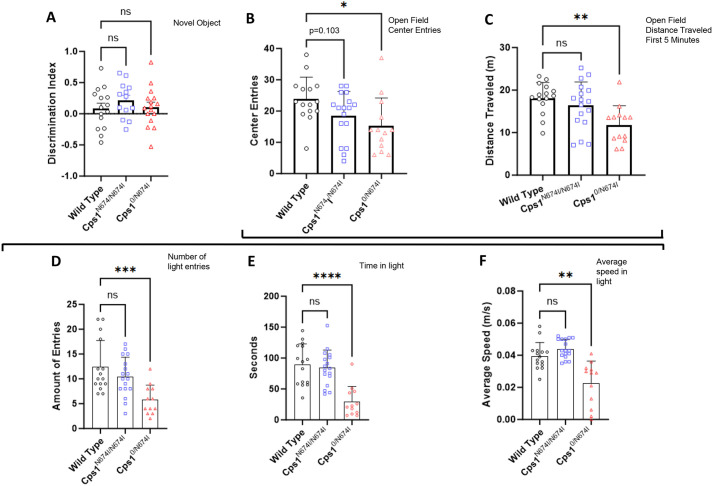
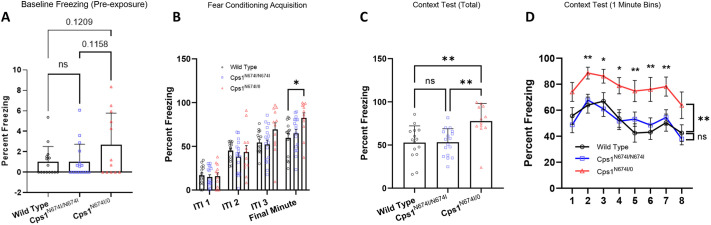
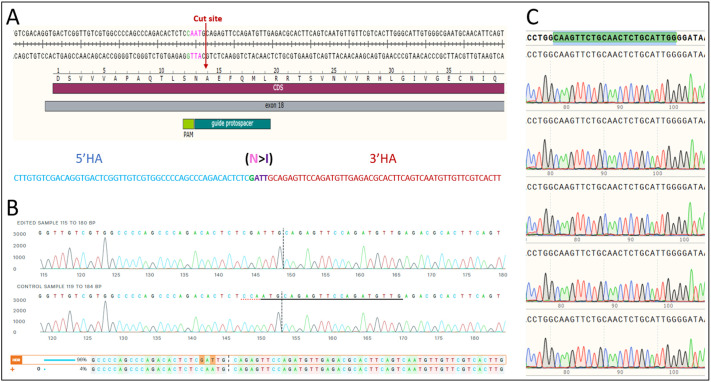
Carbamoyl phosphate synthetase 1 (CPS1) deficiency is a rare metabolic disorder that, in neonatal onset, is typically characterized by severe life-threatening and neurologically injuring hyperammonemic episodes with high unmet patient need. Patients that retain limited enzyme activity may present later in life with less severe hyperammonemia. CPS1 drives the first step in the urea cycle, the pathway terrestrial mammals utilize to metabolize nitrogen. In order to probe the effect of hyperammonemia on the developing nervous system and explore new therapies, a murine Cps1 exon 3-4 mutant was previously generated. However, these mice die within 24 h of birth, limiting study capabilities. Herein, we developed a novel Cps1 hypomorphic murine model with residual enzyme activity that maintains survival, but with dysfunction of Cps1 that could be detected biochemically. Characterization, based on the orthologous human variant Asn674Ile, revealed that the variant is reproducible, 100% penetrant and biochemically phenocopies the human disorder. The hypomorph presents with elevated ammonia and glutamate, and reduced citrulline, and with an impaired rate of ureagenesis, providing a novel platform to study and develop therapies for CPS1 deficiency.
Keywords: Carbamoyl phosphate synthetase 1 deficiency; Enzyme activity; Glutamine; Human mutation; Hyperammonemia.
© 2025. Published by The Company of Biologists.
Conflict of interest statement
Competing interests G.S.L. serves as a consultant to Astellas Gene Therapies and has received grant support from the Association of Creatine Deficiencies in an area unrelated to the work described in this paper. A.M.D. declares the following interests, all of which are in areas unrelated to the work described in this paper: she is a scientific advisor with a financial interest in Fenologica Biosciences, Inc., her laboratory has received grant funding from the National Urea Cycle Disorders Foundation, and her laboratory has received sponsored research funding from Moderna, Inc. All other authors declare no competing or financial interests.
Figures
Similar articles
-
Conditional disruption of hepatic carbamoyl phosphate synthetase 1 in mice results in hyperammonemia without orotic aciduria and can be corrected by liver-directed gene therapy.Mol Genet Metab. 2018 Aug;124(4):243-253. doi: 10.1016/j.ymgme.2018.04.001. Epub 2018 Apr 12. Mol Genet Metab. 2018. PMID: 29801986 Free PMC article.
-
A constitutive knockout of murine carbamoyl phosphate synthetase 1 results in death with marked hyperglutaminemia and hyperammonemia.J Inherit Metab Dis. 2019 Nov;42(6):1044-1053. doi: 10.1002/jimd.12048. Epub 2019 Mar 5. J Inherit Metab Dis. 2019. PMID: 30835861 Free PMC article.
-
Use of an oversized AAV8 vector for CPS1 deficiency results in long-term survival and ammonia control.Mol Ther Nucleic Acids. 2025 Feb 3;36(1):102470. doi: 10.1016/j.omtn.2025.102470. eCollection 2025 Mar 11. Mol Ther Nucleic Acids. 2025. PMID: 40083646 Free PMC article.
-
Genetic, structural and biochemical basis of carbamoyl phosphate synthetase 1 deficiency.Mol Genet Metab. 2010 Dec;101(4):311-23. doi: 10.1016/j.ymgme.2010.08.002. Epub 2010 Aug 6. Mol Genet Metab. 2010. PMID: 20800523 Review.
-
The clinical effectiveness and cost-effectiveness of enzyme replacement therapy for Gaucher's disease: a systematic review.Health Technol Assess. 2006 Jul;10(24):iii-iv, ix-136. doi: 10.3310/hta10240. Health Technol Assess. 2006. PMID: 16796930
References
-
- Allegri, G., Deplazes, S., Grisch-Chan, H. M., Mathis, D., Fingerhut, R., Haberle, J. and Thony, B. (2017). A simple dried blood spot-method for in vivo measurement of ureagenesis by gas chromatography-mass spectrometry using stable isotopes. Clin. Chim. Acta 464, 236-243. 10.1016/j.cca.2016.11.038 - DOI - PubMed
-
- Allegri, G., Deplazes, S., Rimann, N., Causton, B., Scherer, T., Leff, J. W., Diez-Fernandez, C., Klimovskaia, A., Fingerhut, R., Krijt, J.et al. (2019). Comprehensive characterization of ureagenesis in the spf(ash) mouse, a model of human ornithine transcarbamylase deficiency, reveals age-dependency of ammonia detoxification. J. Inherit. Metab. Dis. 42, 1064-1076. 10.1002/jimd.12068 - DOI - PubMed
MeSH terms
Substances
Grants and funding
- 5P50HD103557/National Institute of Child Health and Human Development
- R01HD114863/National Institute of Child Health and Human Development
- 320030_207965/Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung
- P50 HD103557/HD/NICHD NIH HHS/United States
- University of California, Los Angeles
LinkOut - more resources
Full Text Sources
