

mTOR pathway diseases: challenges and opportunities from bench to bedside and the mTOR node
- PMID: 40426219
- PMCID: PMC12107773
- DOI: 10.1186/s13023-025-03740-1
mTOR pathway diseases: challenges and opportunities from bench to bedside and the mTOR node
Abstract
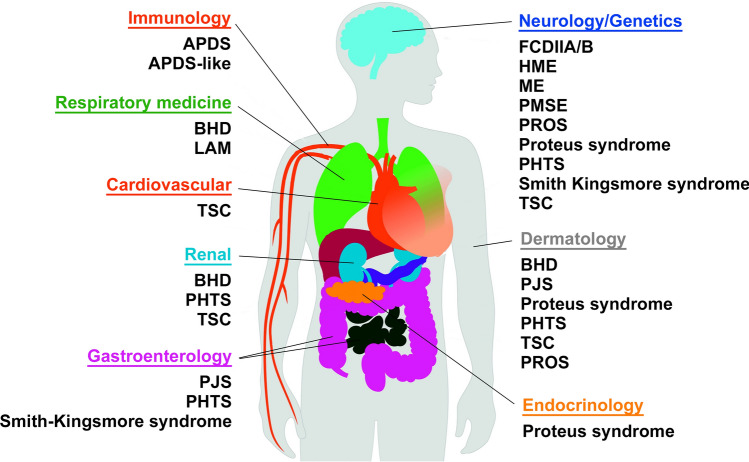
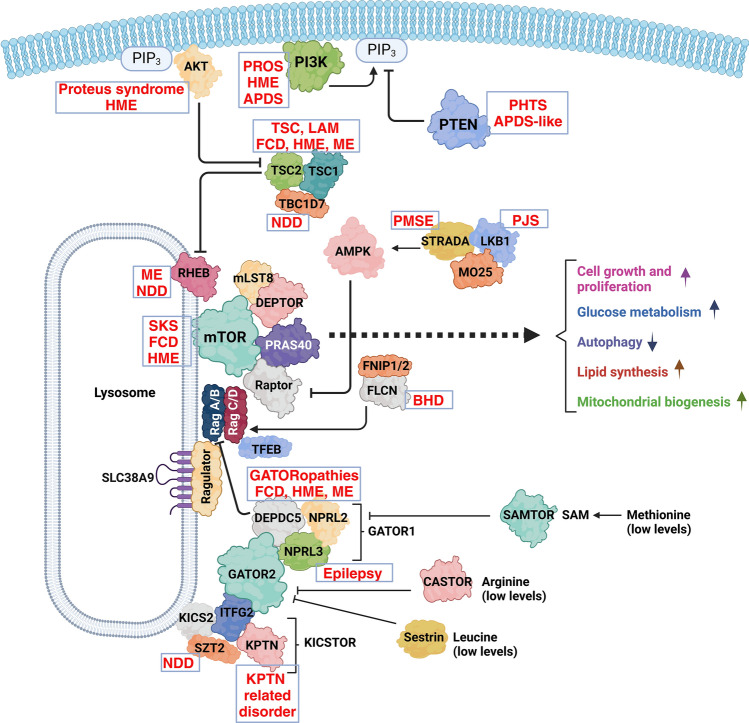
Mechanistic target of rapamycin (mTOR) is a highly conserved serine/threonine kinase that regulates key cellular processes including cell growth, autophagy and metabolism. Hyperactivation of the mTOR pathway causes a group of rare and ultrarare genetic diseases. mTOR pathway diseases have diverse clinical manifestations that are managed by distinct medical disciplines but share a common underlying molecular basis. There is a now a deep understanding of the molecular underpinning that regulates the mTOR pathway but effective treatments for most mTOR pathway diseases are lacking. Translating scientific knowledge into clinical applications to benefit the unmet clinical needs of patients is a major challenge common to many rare diseases. In this article we expound how mTOR pathway diseases provide an opportunity to coordinate basic and translational disease research across the group, together with industry, medical research foundations, charities and patient groups, by pooling expertise and driving progress to benefit patients. We outline the germline and somatic mutations in the mTOR pathway that cause rare diseases and summarise the prevalence, genetic basis, clinical manifestations, pathophysiology and current treatments for each disease in this group. We describe the challenges and opportunities for progress in elucidating the underlying mechanisms, improving diagnosis and prognosis, as well as the development and approval of new therapies for mTOR pathway diseases. We illustrate the crucial role of patient public involvement and engagement in rare disease and mTOR pathway disease research. Finally, we explain how the mTOR Pathway Diseases node, part of the Research Disease Research UK Platform, will address these challenges to improve the understanding, diagnosis and treatment of mTOR pathway diseases.
Keywords: AKT; Birt-Hogg-Dubé; Everolimus; PI3K; PTEN; Peutz-Jeghers; Rapamycin; Rare diseases; Tuberous sclerosis complex; mTOR.
© 2025. The Author(s).
Conflict of interest statement
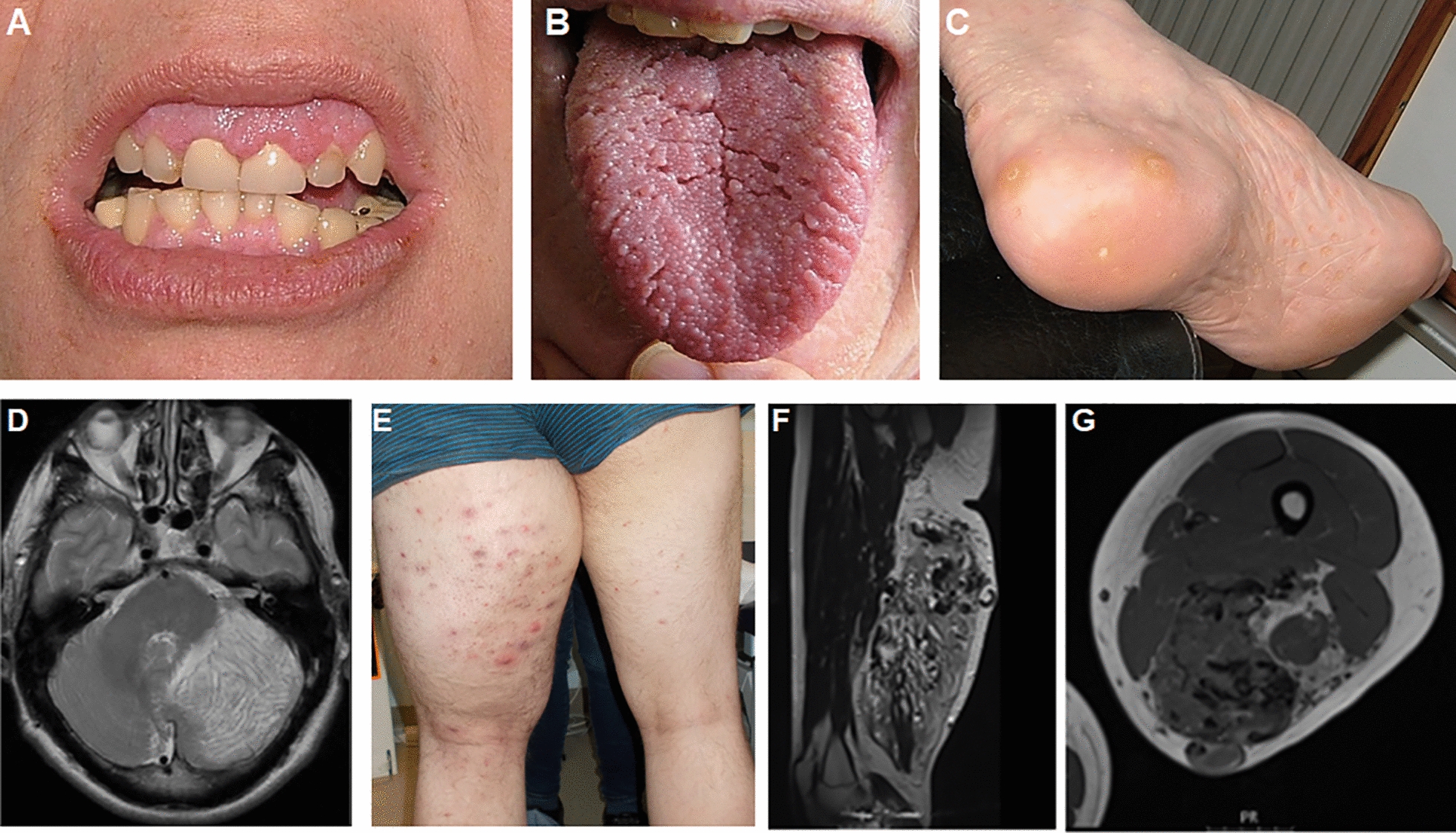
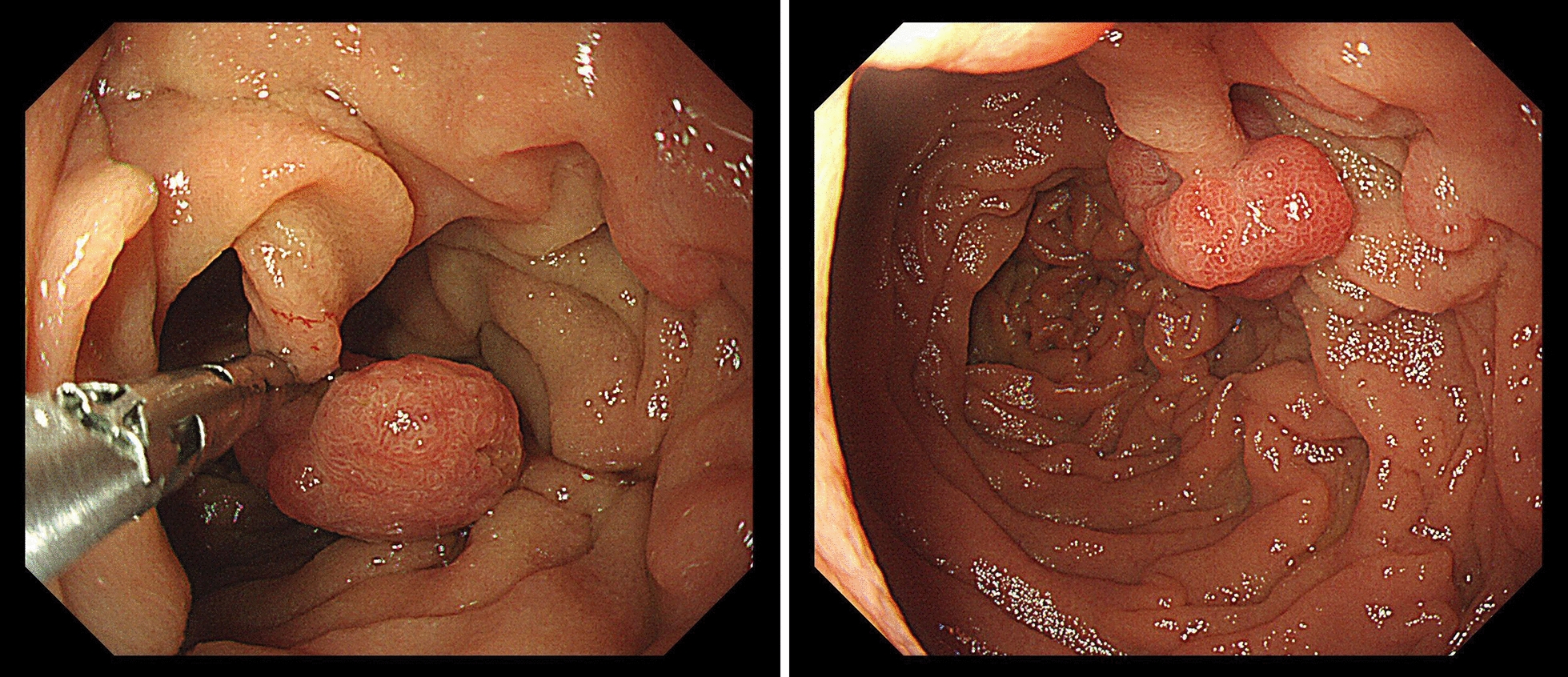
Declarations. Ethics approval and consent to participate: Patient data were recorded as part of routine clinical practice and not as part of a research study so do not require ethical approval, apart from data in Fig. 9 which was part of a study approved by the Wales REC committee REC number 22/WA/0326. Consent for publication: Consent for publication was given for all patient related data. Competing interests: SD is a UCB employee and SD receives stock or stock options from their employment. SH has received consultancy fees and research funding from Pharming in the past. DP and JMB are members of the scientific advisory board for CoSyne.
Figures
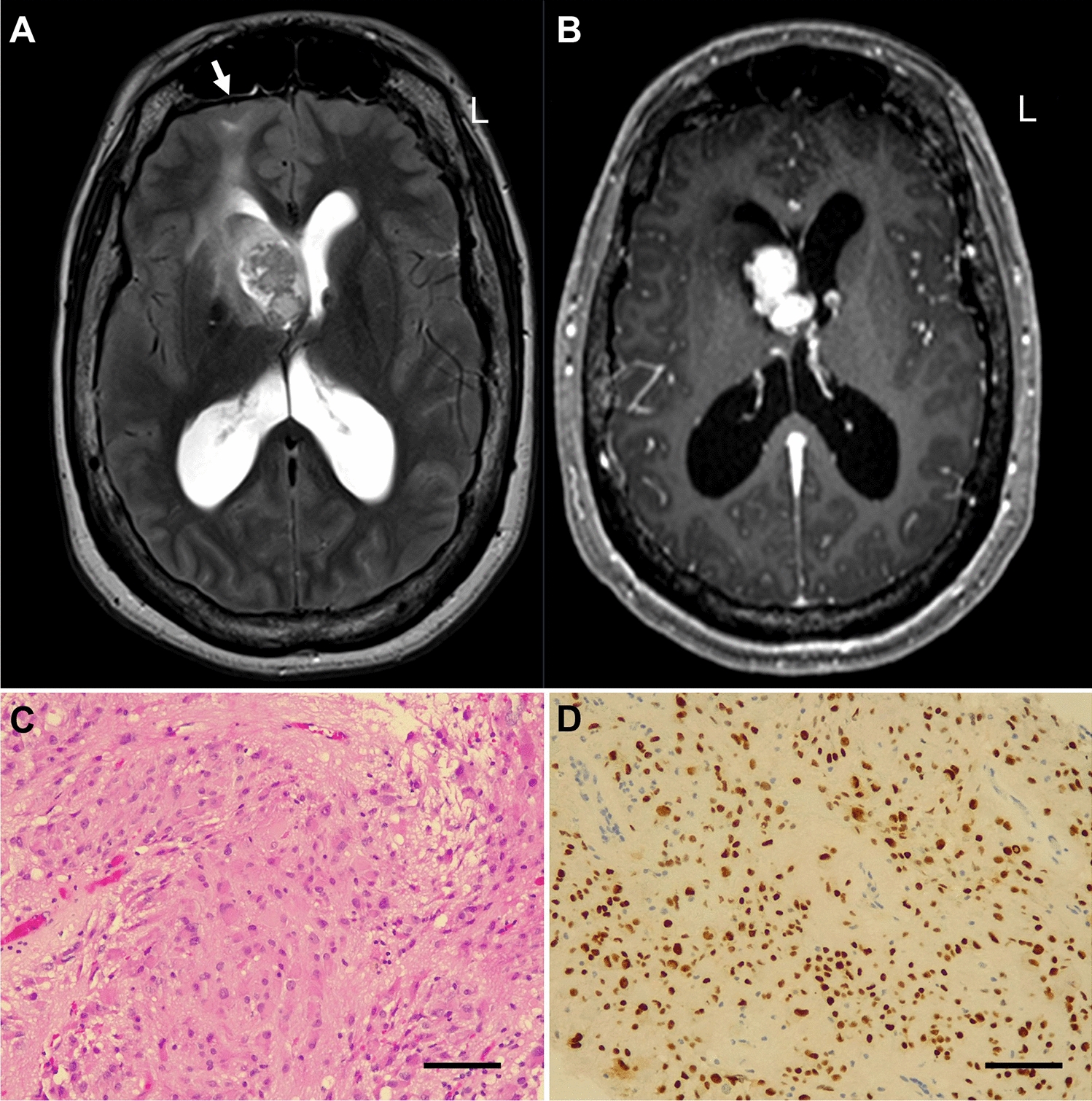
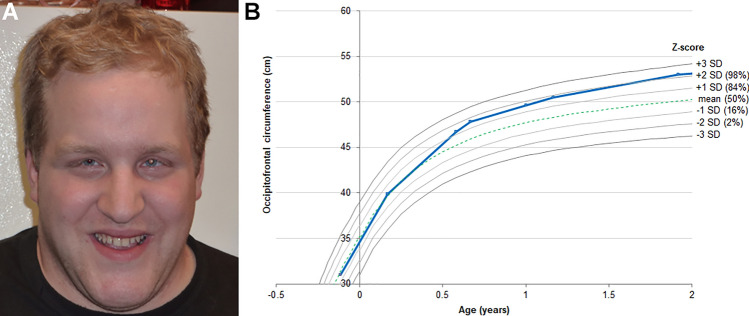
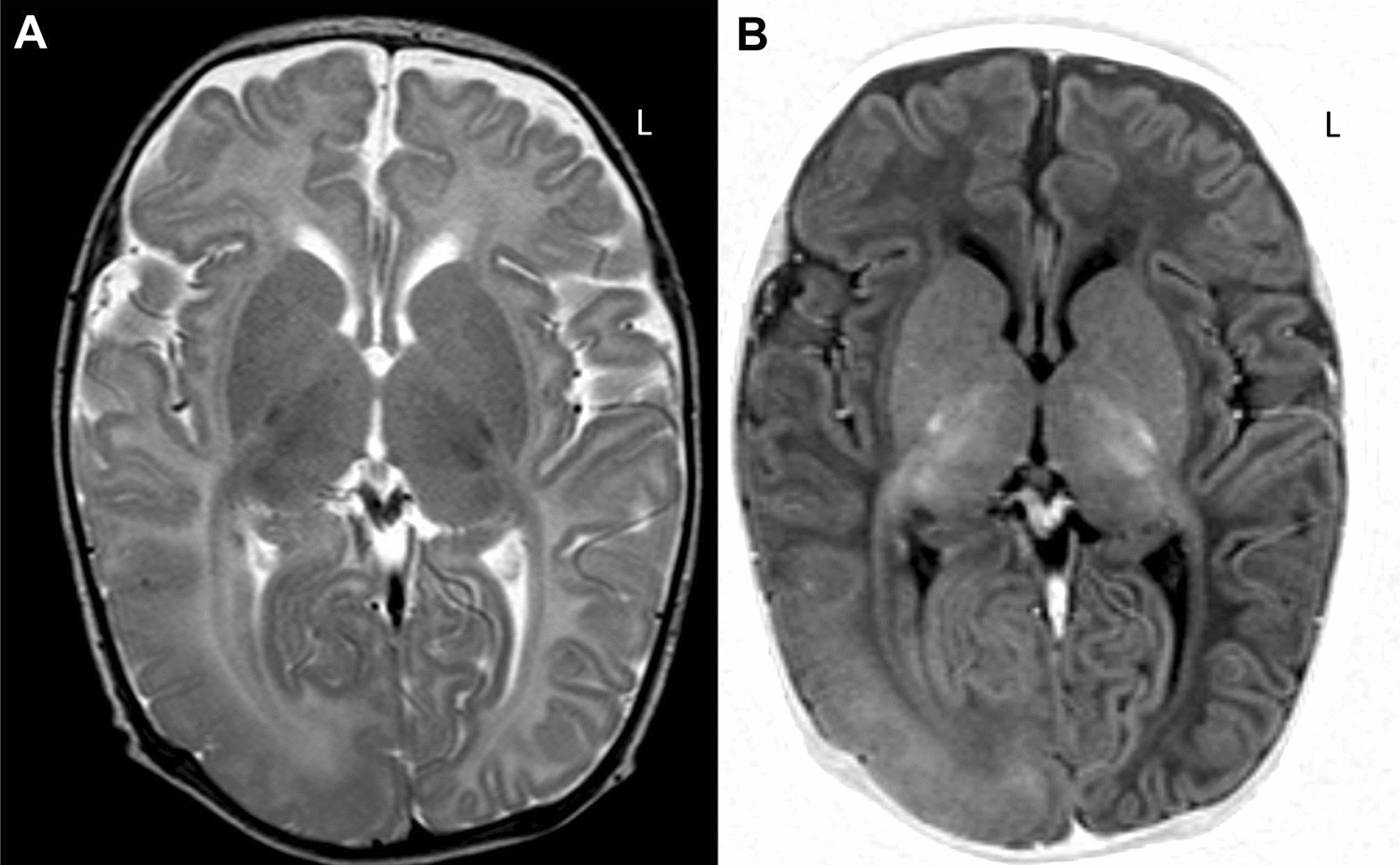
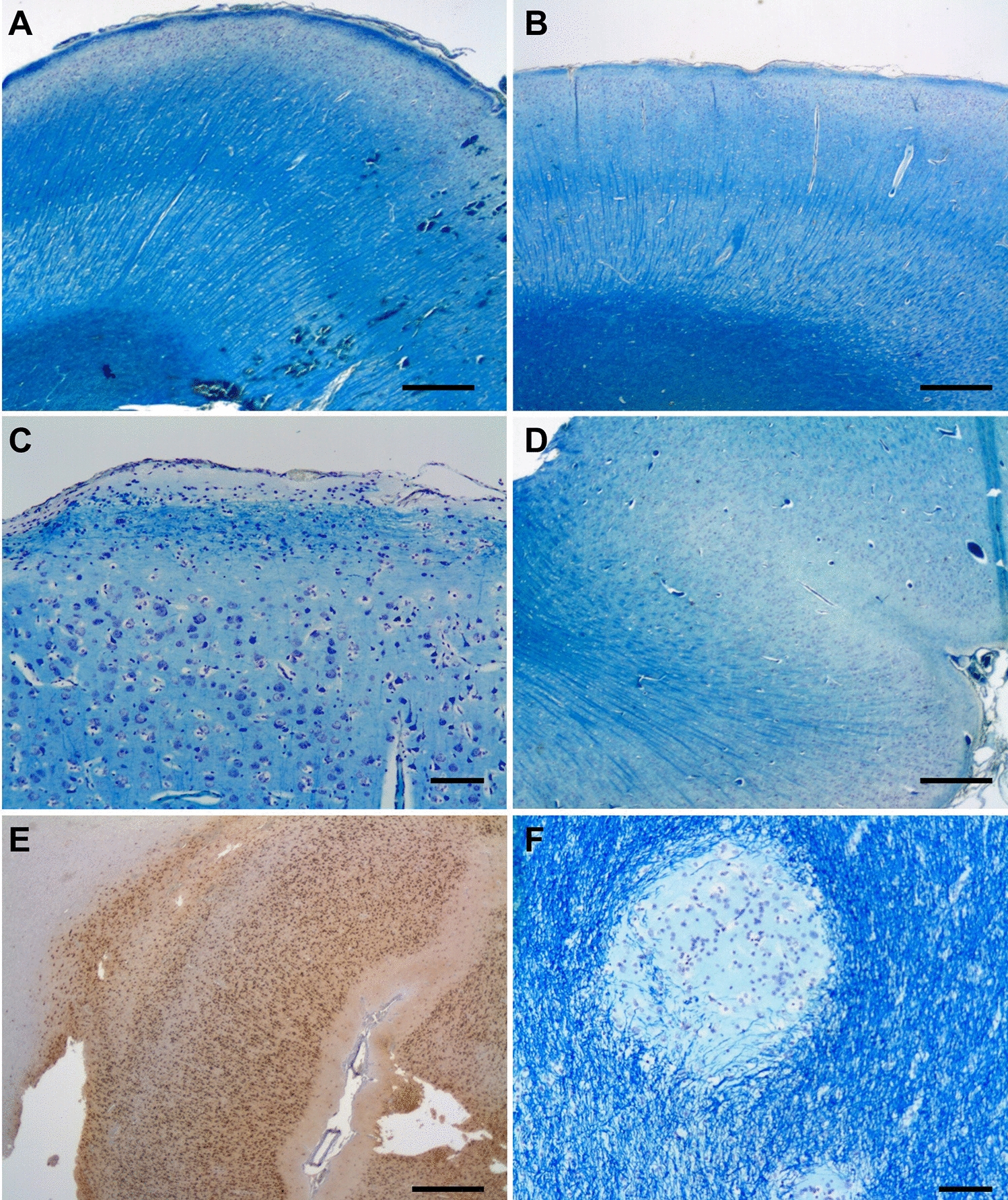
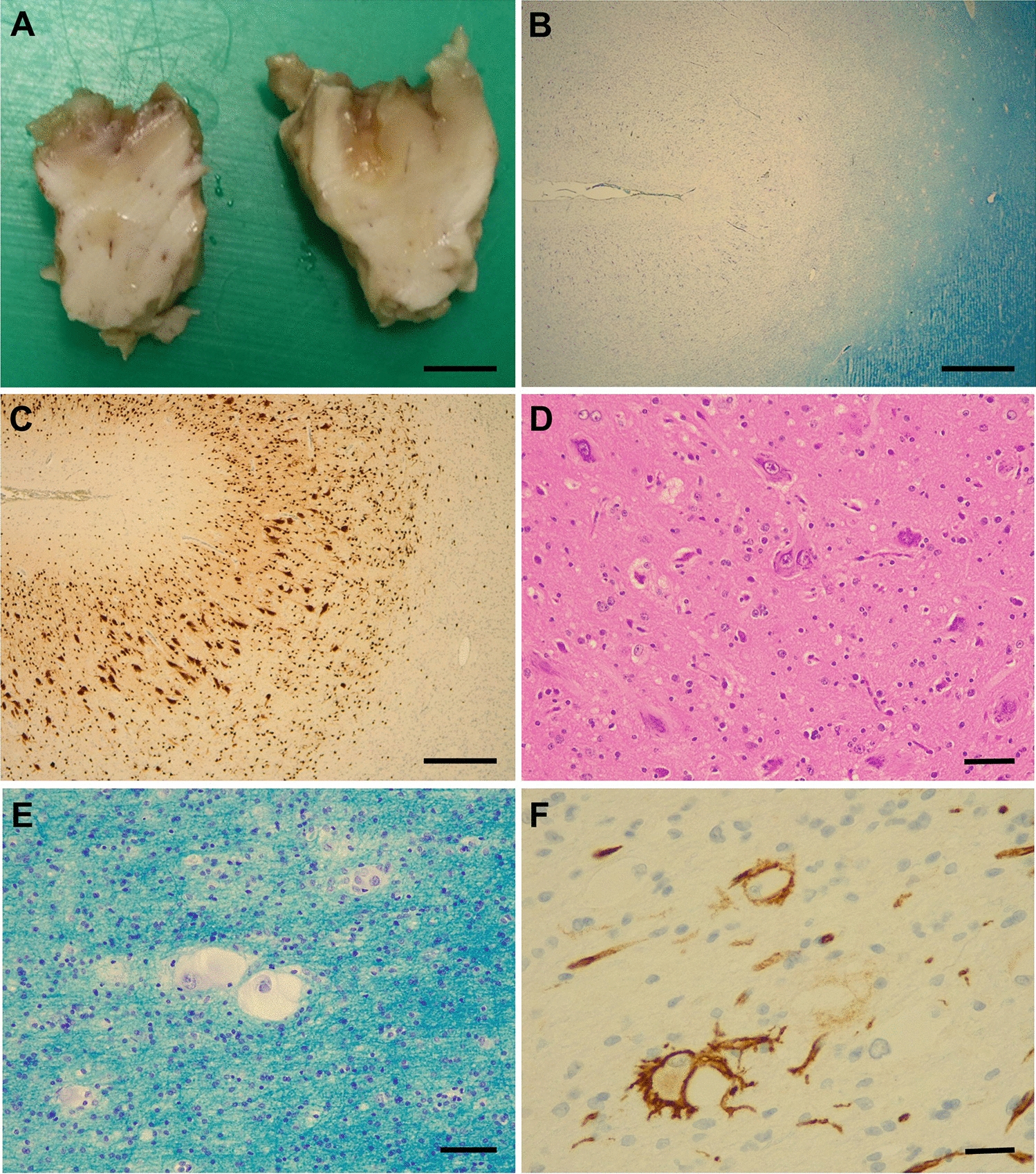
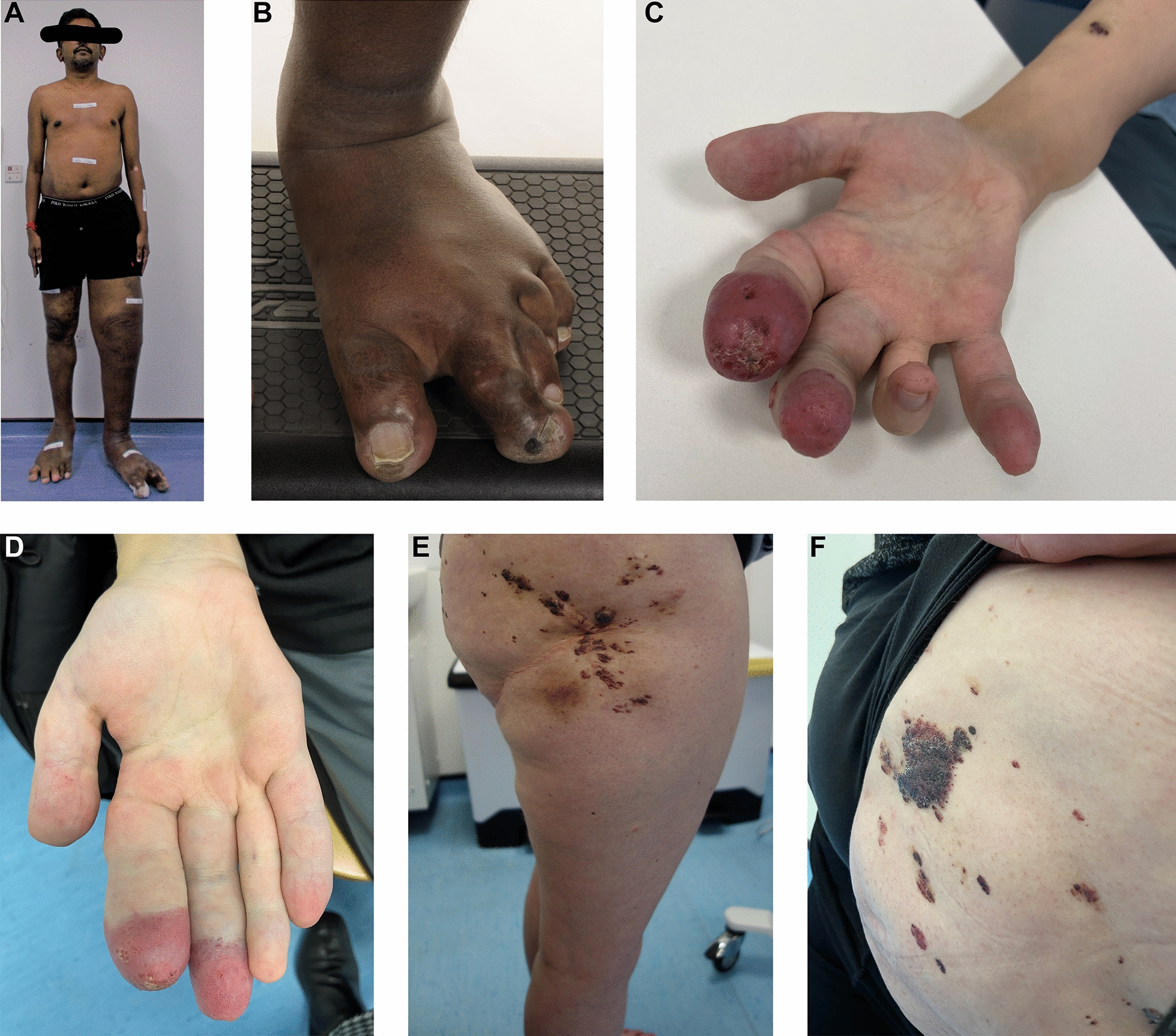
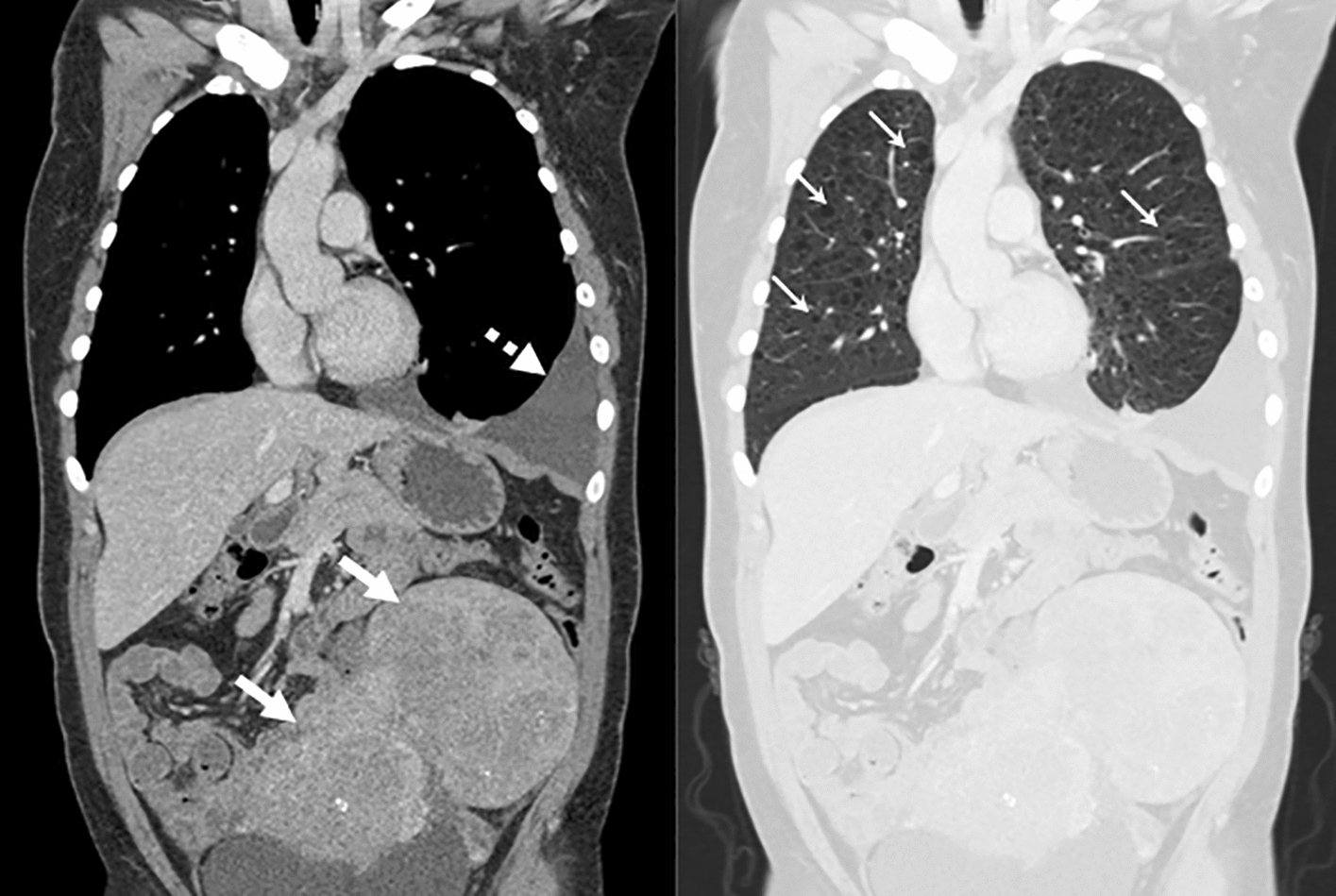
References
-
- O’Connor DJ, Gabaldo M, Aartsma-Rus A, Hechtelt JA. Defining rare conditions in the era of personalized medicine. Nat Rev Drug Discov. 2023;22(11):857–8. - PubMed
-
- UK Rare Diseases Framework [Available from: https://www.gov.uk/government/publications/uk-rare-diseases-framework. Accessed on 30th Sept 2024
-
- mTOR Pathway Diseases [Available from: https://rd-research.org.uk/node/mtor-pathway-diseases/. Accessed on 30th Sept 2024
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
