

Identification and Functional Characterization of a Novel SOX4 Mutation Predisposing to Coffin-Siris Syndromic Congenital Heart Disease
- PMID: 40426787
- PMCID: PMC12109673
- DOI: 10.3390/children12050608
Identification and Functional Characterization of a Novel SOX4 Mutation Predisposing to Coffin-Siris Syndromic Congenital Heart Disease
Abstract
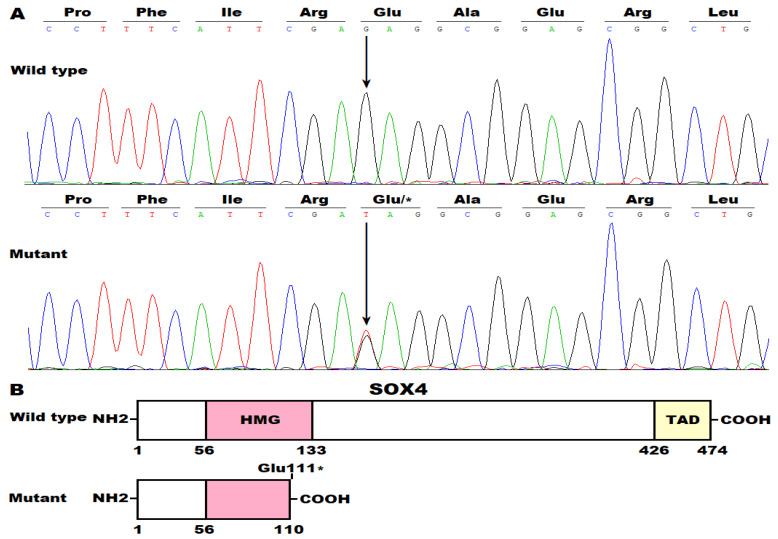
Background/Objectives: Congenital heart disease (CHD) occurs in ~1% of all live neonates globally, rendering it the most prevalent developmental anomaly affecting humans; this condition confers substantial infant morbidity and mortality worldwide. Although there is ample evidence to suggest a paramount genetic basis for CHD, the genetic etiologies underpinning the majority of CHD remain elusive. In the present study, SOX4 was selected as a significant candidate gene for human CHD, mainly because SOX4 is abundantly expressed in both human and murine hearts during embryogenesis, and the knockout of Sox4 in mice causes embryonic demise predominantly attributable to cardiovascular developmental defects. Methods: Sequencing analysis of SOX4 was fulfilled in 248 probands affected with various types of CHD and the available relatives of the identified variation carrier as well as 262 unrelated healthy individuals. Functional analysis of the mutant SOX4 protein was conducted by utilizing a dual-reporter gene system. Results: a novel heterozygous SOX4 variation, NM_003107.3:c.331G>T;p.(Glu111*), was discovered in a male proband with Coffin-Siris syndromic CHD. Genetic investigation of the proband's available relatives revealed that the truncating variation co-segregated with the phenotype in the whole family. The nonsense variation was absent from 262 healthy controls. Functional analysis demonstrated that the Glu111*-mutant SOX4 lost transactivation on NKX2.5 and GATA4, two well-established genes that are causative factors for CHD. Moreover, the Glu111* mutation nullified the synergistic transactivation between SOX4 and TBX20, another CHD-causing gene. Conclusions: These findings support SOX4 as a causative gene accountable for familial Coffin-Siris syndromic CHD in humans. These findings may aid in developing personalized preventive and therapeutic strategies for patients with Coffin-Siris syndromic CHD.
Keywords: SOX4; biological assay; congenital heart disease; human molecular genetics; patent ductus arteriosus; transcription regulation.
Conflict of interest statement
The authors declare that no conflict of interest exists.
Figures




References
-
- Martin S.S., Aday A.W., Almarzooq Z.I., Anderson C.A.M., Arora P., Avery C.L., Baker-Smith C.M., Gibbs B., Beaton A.Z., Boehme A.K., et al. 2024 Heart Disease and Stroke Statistics: A Report of US and Global Data From the American Heart Association. Circulation. 2024;149:e347–e913. doi: 10.1161/CIR.0000000000001209. - DOI - PMC - PubMed
-
- Ly R., Karsenty C., Amedro P., Cohen S., Domanski O., Godart F., Radojevic J., Vaksmann G., Naccache N., Boubrit A., et al. Health-Related Quality of Life and Its Association With Outcomes in Adults With Congenital Heart Disease and Heart Failure: Insight From FRESH-ACHD Registry. J. Am. Heart Assoc. 2023;12:e027819. doi: 10.1161/JAHA.122.027819. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
