

Genotype-Phenotype Associations in Phelan-McDermid Syndrome: Insights into Novel Genes Beyond SHANK3
- PMID: 40429797
- PMCID: PMC12111097
- DOI: 10.3390/ijms26104653
Genotype-Phenotype Associations in Phelan-McDermid Syndrome: Insights into Novel Genes Beyond SHANK3
Abstract
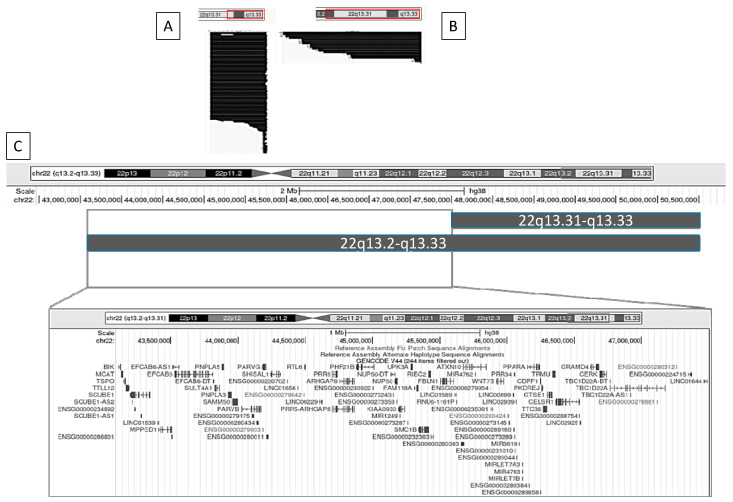
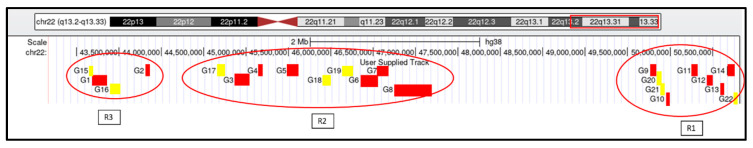
Phelan-McDermid syndrome (PMS; #MIM: 606232) is a rare neurodevelopmental disorder primarily caused by the haploinsufficiency of the SHANK3 gene, most often due to deletions encompassing the gene or single nucleotide variants within it. Individuals with PMS display a wide range of clinical abnormalities and considerable genetic heterogeneity. This study aims to investigate genotype-phenotype correlations in a cohort of 213 individuals with PMS and to identify novel candidate genes, beyond SHANK3, that may contribute to the syndrome's diverse clinical manifestations. Unsupervised clustering based on deletion size and Global Functional Assessment of the Patient (GFAP, previously described and developed by our group), along with additional analytical approaches, were employed to explore genotype-phenotype relationships. Deletion size within the 22q13.3 region emerged as a major determinant of phenotype, with larger deletions associated with more severe global functional impairment. Furthermore, CERK, TBC1D22A, CELSR1, and GRAMD4 were identified as candidate genes within 22q13.3, potentially contributing to core PMS phenotypes, and their putative interactions were explored. Our findings support the central role of SHANK3 in PMS, while also indicating that it does not account for the full phenotypic spectrum. This study underscores the variable impact of distinct genetic alterations in PMS and proposes additional loci implicated in its pathogenesis. These insights may inform future therapeutic strategies, emphasizing the importance of patient stratification and precision medicine.
Keywords: 22q13.33 chromosomal region; Phelan–McDermid syndrome (PMS); SHANK3; genotype–phenotype correlation; haploinsufficiency.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures






References
-
- Schön M., Lapunzina P., Nevado J., Mattina T., Gunnarsson C., Hadzsiev K., Verpelli C., Bourgeron T., Jesse S., van Ravenswaaij-Arts C.M.A., et al. Definition and clinical variability of SHANK3-related Phelan-McDermid syndrome. Eur. J. Med. Genet. 2023;66:104754. doi: 10.1016/j.ejmg.2023.104754. - DOI - PubMed
-
- Nevado J., García-Miñaúr S., Palomares-Bralo M., Vallespín E., Guillén-Navarro E., Rosell J., Bel-Fenellós C., Mori M.Á., Milá M., Campo M.D., et al. Variability in Phelan-McDermid Syndrome in a Cohort of 210 Individuals. Front. Genet. 2022;13:652454. doi: 10.3389/fgene.2022.652454. - DOI - PMC - PubMed
-
- Levy T., Foss-Feig J.H., Betancur C., Siper P.M., Trelles-Thorne M.d.P., Halpern D., Frank Y., Lozano R., Layton C., Britvan B., et al. Strong evidence for genotype–Phenotype correlations in Phelan-McDermid syndrome: Results from the developmental synaptopathies consortium. Hum. Mol. Genet. 2022;31:625–637. doi: 10.1093/hmg/ddab280. - DOI - PMC - PubMed
-
- De Rubeis S., Siper P.M., Durkin A., Weissman J., Muratet F., Halpern D., Trelles M.P., Frank Y., Lozano R., Ting Wang A., et al. Delineation of the Genetic and Clinical Spectrum of Phelan-McDermid Syndrome Caused by SHANK3 point Mutations. Mol. Autism. 2018;9:31. doi: 10.1186/s13229-018-0205-9. - DOI - PMC - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Miscellaneous
