

Alphaflexiviridae in Focus: Genomic Signatures, Conserved Elements and Viral-Driven Cellular Remodeling
- PMID: 40431623
- PMCID: PMC12115993
- DOI: 10.3390/v17050611
Alphaflexiviridae in Focus: Genomic Signatures, Conserved Elements and Viral-Driven Cellular Remodeling
Abstract
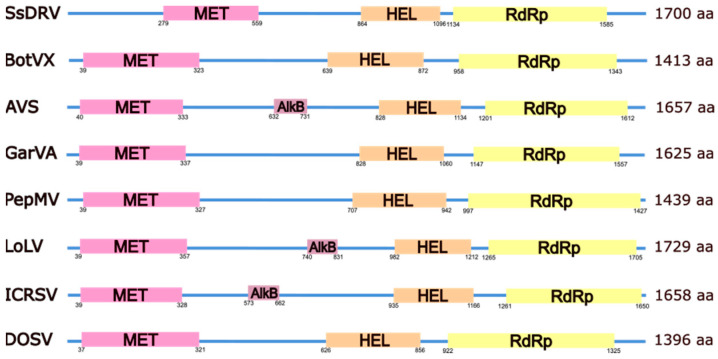
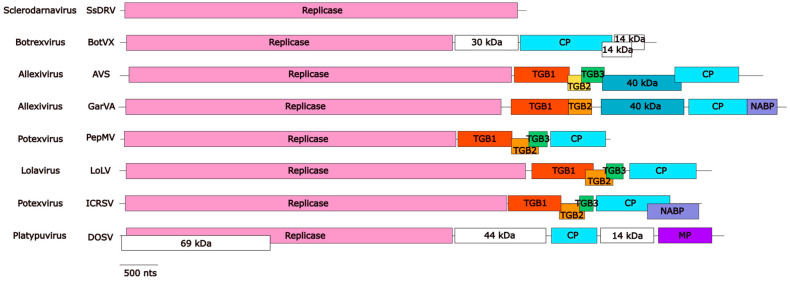
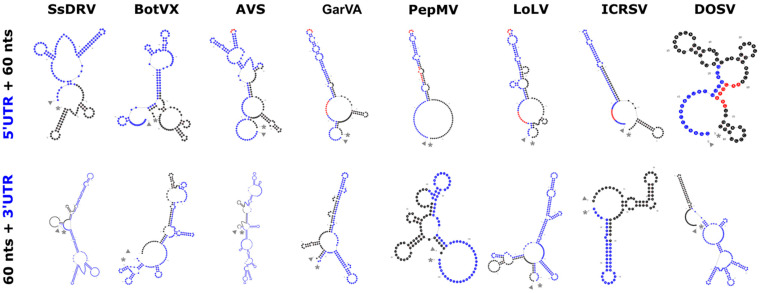
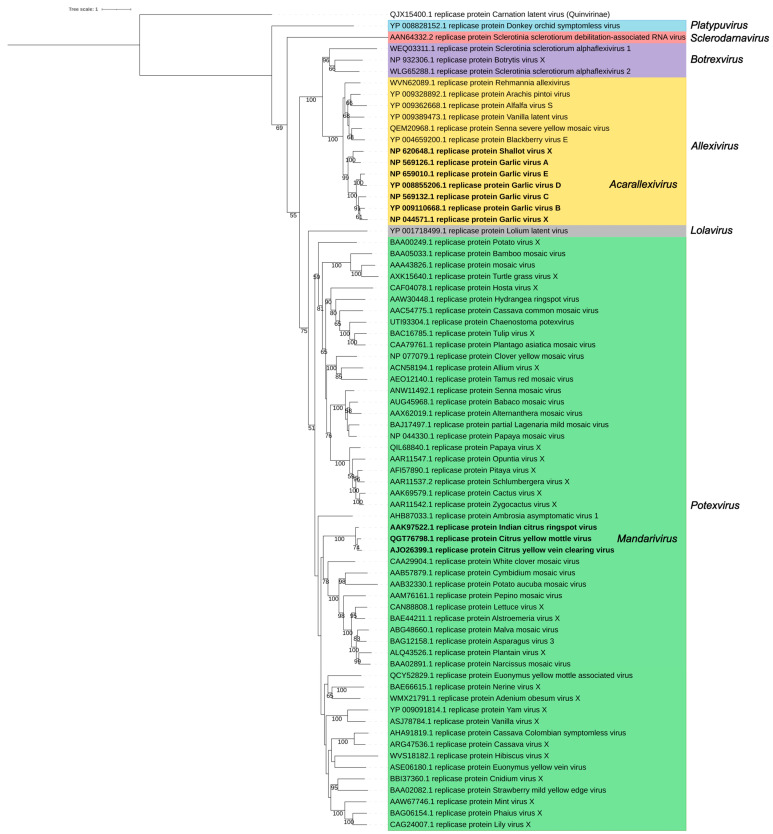
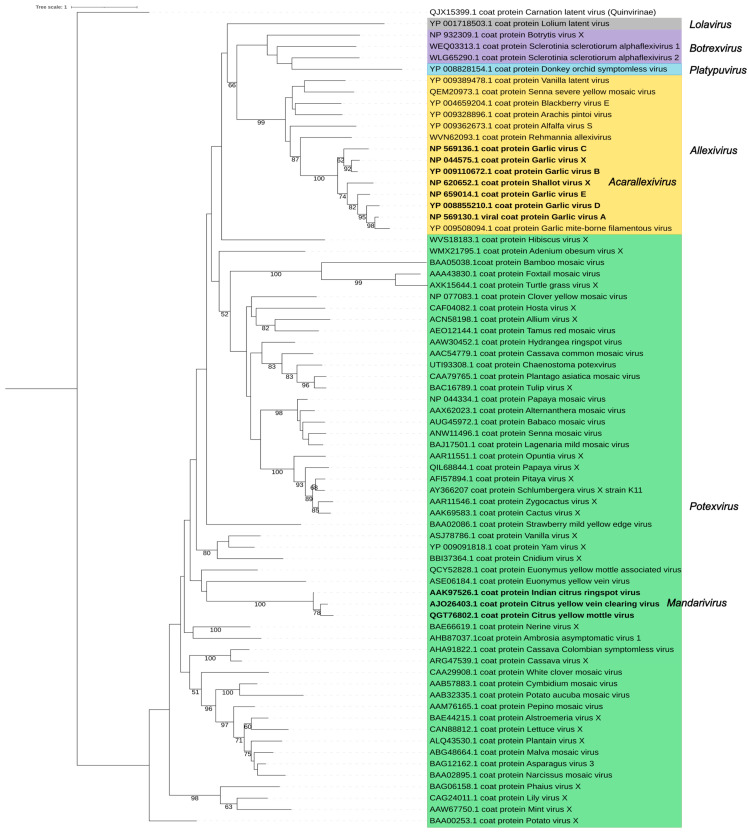
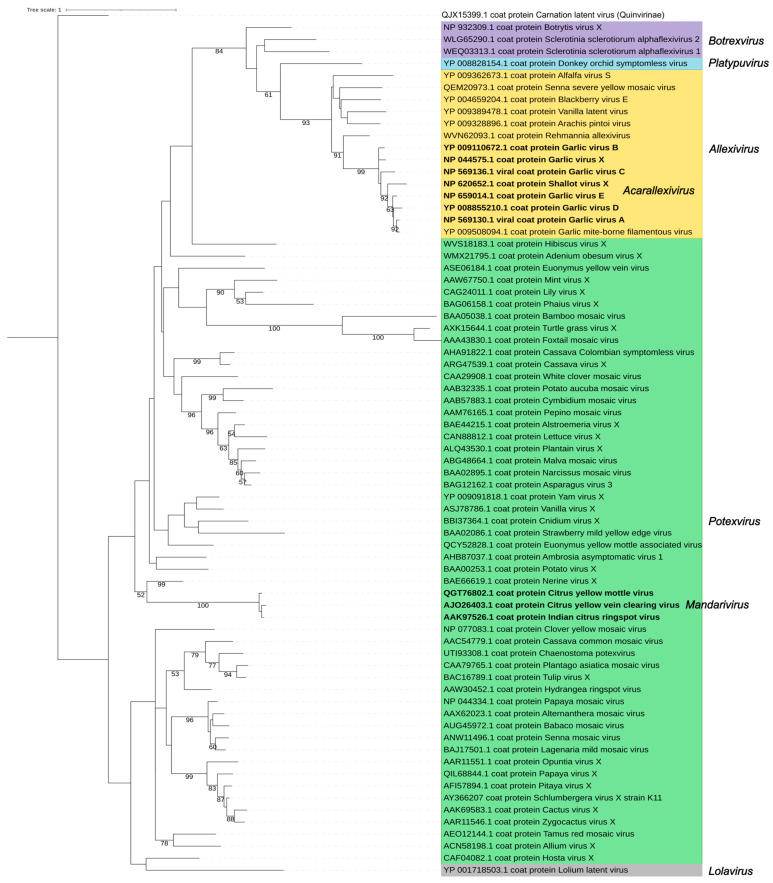
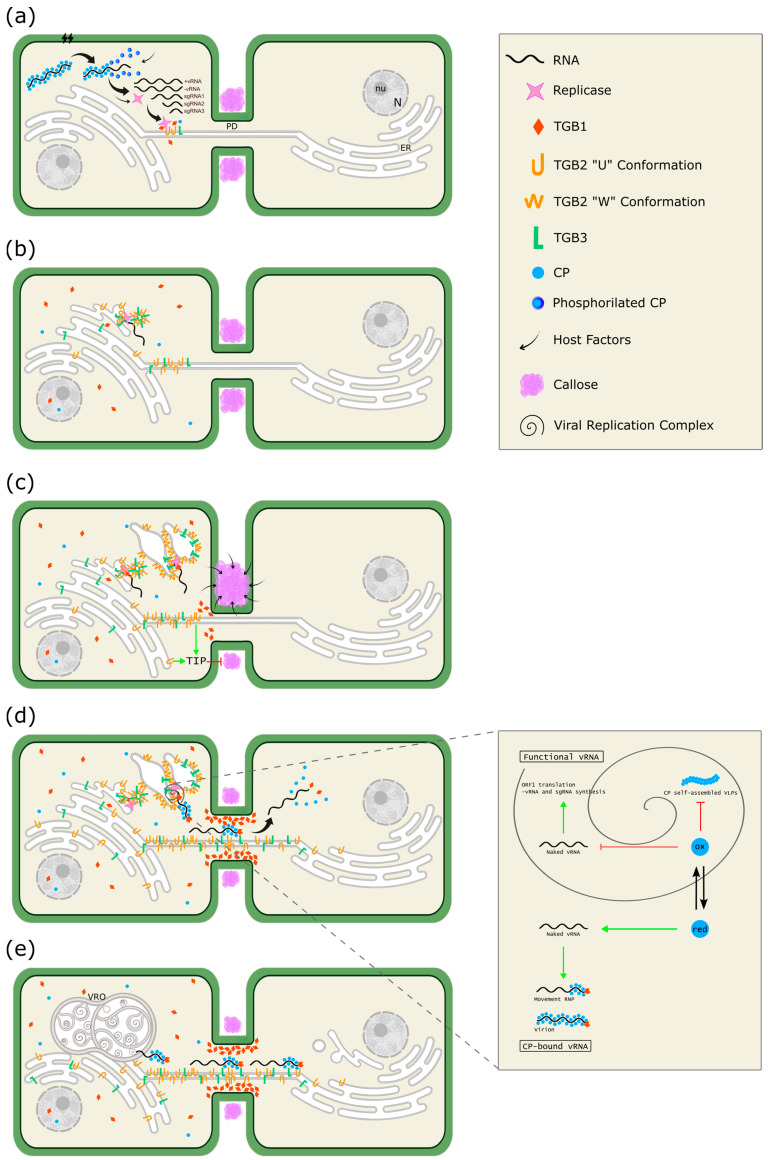
The family Alphaflexiviridae comprises plant- and fungus-infecting viruses with single-stranded, positive-sense RNA genomes ranging from 5.4 to 9 kb. Their virions are flexuous and filamentous, measuring 470-800 nm in length and 12-13 nm in diameter. The family includes 72 recognized species, classified into six genera: Allexivirus, Lolavirus, Platypuvirus, Potexvirus (plant-infecting), and Botrexvirus and Sclerodarnavirus (fungus-infecting). The genus Potexvirus is the largest, with 52 species, including Potexvirus ecspotati (potato virus X), an important crop pathogen and plant virology model. The genera are distinguished by genome organization and host range, while species differentiation relies on nucleotide and protein sequence identity thresholds. In this review, we summarize the current knowledge on the genomic structure, conserved genes, and phylogenetic relationships within Alphaflexiviridae, with a particular focus on the replicase and coat protein genes as signature markers. Additionally, we update the model of cellular remodeling driven by the triple gene block proteins, which are essential for virus movement, among other viral functions. Beyond their biological significance, alphaflexiviruses serve as valuable models for studying virus-host dynamics and hold potential applications in plant disease control and biotechnology. This review provides an updated framework for understanding Alphaflexiviridae and their broader impact on plant virology.
Keywords: Allexivirus; Botrexvirus; Lolavirus; Platypuvirus; Potexvirus; Sclerodarnavirus; coat protein; replicase; triple gene block proteins.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures

Similar articles
-
Identification of nine putative novel members of plant-infecting alphaflexiviruses in public domain plant transcriptomes.Virusdisease. 2024 Dec;35(4):630-636. doi: 10.1007/s13337-024-00898-3. Epub 2024 Oct 19. Virusdisease. 2024. PMID: 39677843
-
Novel polymycoviruses are encapsidated in filamentous virions.J Virol. 2025 Jan 31;99(1):e0151524. doi: 10.1128/jvi.01515-24. Epub 2024 Dec 10. J Virol. 2025. PMID: 39655956 Free PMC article.
-
ICTV Virus Taxonomy Profile: Alphaflexiviridae.J Gen Virol. 2020 Jul;101(7):699-700. doi: 10.1099/jgv.0.001436. Epub 2020 Jun 4. J Gen Virol. 2020. PMID: 32525472 Free PMC article.
-
Family Flexiviridae: a case study in virion and genome plasticity.Annu Rev Phytopathol. 2007;45:73-100. doi: 10.1146/annurev.phyto.45.062806.094401. Annu Rev Phytopathol. 2007. PMID: 17362202 Review.
-
On the Trail of the Longest Plant RNA Virus: Citrus Tristeza Virus.Viruses. 2025 Mar 31;17(4):508. doi: 10.3390/v17040508. Viruses. 2025. PMID: 40284951 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
