

Domesticated cannabinoid synthases amid a wild mosaic cannabis pangenome
- PMID: 40437092
- PMCID: PMC12286863
- DOI: 10.1038/s41586-025-09065-0
Domesticated cannabinoid synthases amid a wild mosaic cannabis pangenome
Abstract
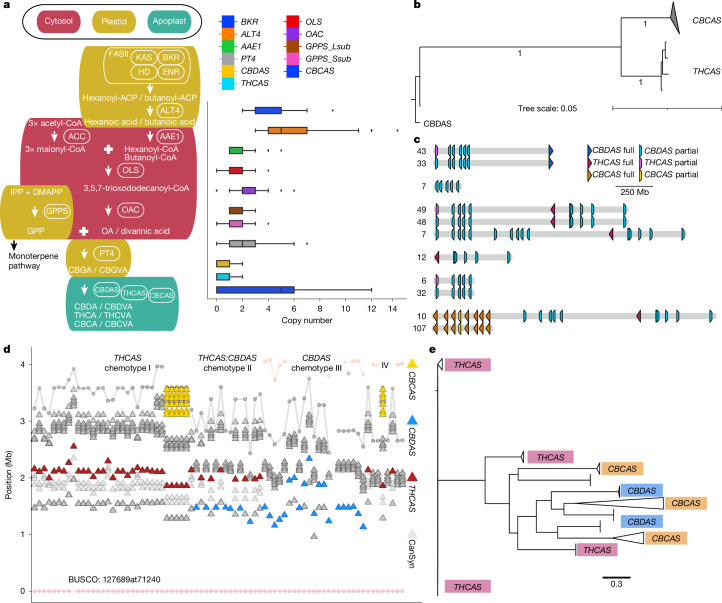
Cannabis sativa is a globally important seed oil, fibre and drug-producing plant species. However, a century of prohibition has severely restricted development of breeding and germplasm resources, leaving potential hemp-based nutritional and fibre applications unrealized. Here we present a cannabis pangenome, constructed with 181 new and 12 previously released genomes from a total of 144 biological samples including both male (XY) and female (XX) plants. We identified widespread regions of the cannabis pangenome that are surprisingly diverse for a single species, with high levels of genetic and structural variation, and propose a novel population structure and hybridization history. Across the ancient heteromorphic X and Y sex chromosomes, we observed a variable boundary at the sex-determining and pseudoautosomal regions as well as genes that exhibit male-biased expression, including genes encoding several key flowering regulators. Conversely, the cannabinoid synthase genes, which are responsible for producing cannabidiol acid and delta-9-tetrahydrocannabinolic acid, contained very low levels of diversity, despite being embedded within a variable region with multiple pseudogenized paralogues, structural variation and distinct transposable element arrangements. Additionally, we identified variants of acyl-lipid thioesterase genes that were associated with fatty acid chain length variation and the production of the rare cannabinoids, tetrahydrocannabivarin and cannabidivarin. We conclude that the C. sativa gene pool remains only partially characterized, the existence of wild relatives in Asia is likely and its potential as a crop species remains largely unrealized.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: S.C. was a co-founder of Oregon CBD. A.R.G. and A.T. were employees of Oregon CBD. R.C.L. is a stakeholder in Saint Vrain Research LLC, which manufactures hemp-based products. T.P.M. is a founder of the carbon sequestration company CQuesta. A.H. is a co-founder of the genotyping company Veil Genomics. The other authors declare no competing interests.
Figures














References
-
- Long, T., Wagner, M., Demske, D., Leipe, C. & Tarasov, P. E. Cannabis in Eurasia: origin of human use and Bronze Age trans-continental connections. Veg. Hist. Archaeobot.26, 245–258 (2017).
-
- Bai, Y. et al. Archaeobotanical evidence of the use of medicinal cannabis in a secular context unearthed from south China. J. Ethnopharmacol.275, 114114 (2021). - PubMed
-
- Kovalchuk, I. et al. The genomics of cannabis and its close relatives. Annu. Rev. Plant Biol.71, 713–739 (2020). - PubMed
-
- Clarke, R. & Merlin, M. Cannabis: Evolution and Ethnobotany (Univ. of California Press, 2016).
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
