

SAVANA: reliable analysis of somatic structural variants and copy number aberrations using long-read sequencing
- PMID: 40437218
- PMCID: PMC12240814
- DOI: 10.1038/s41592-025-02708-0
SAVANA: reliable analysis of somatic structural variants and copy number aberrations using long-read sequencing
Abstract
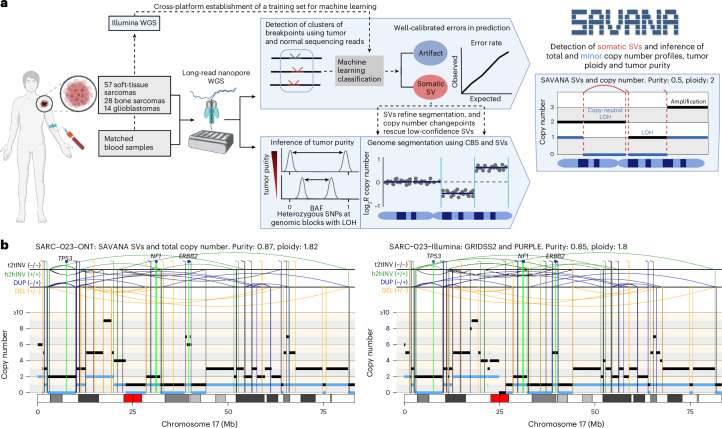
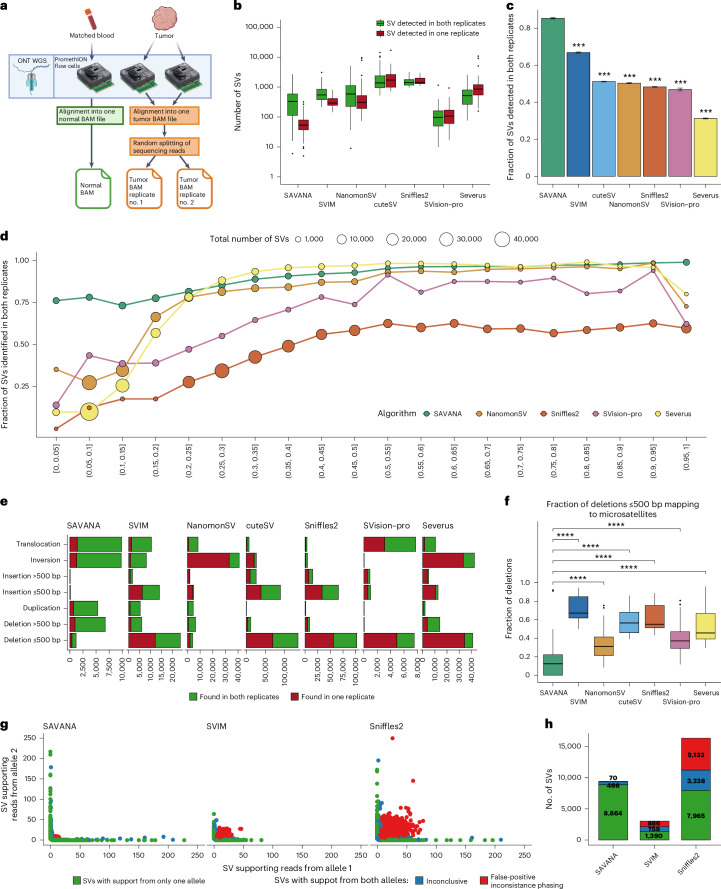
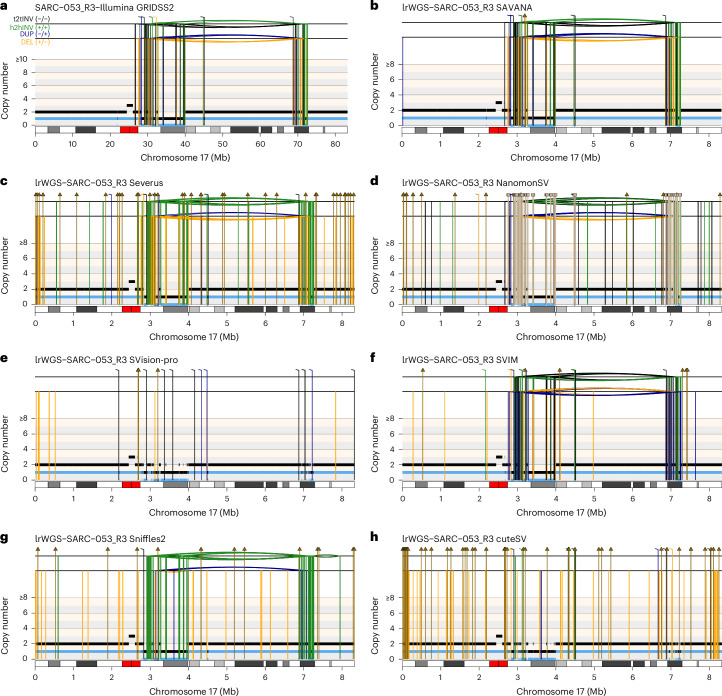
Accurate detection of somatic structural variants (SVs) and somatic copy number aberrations (SCNAs) is critical to study the mutational processes underpinning cancer evolution. Here we describe SAVANA, an algorithm designed to detect somatic SVs and SCNAs at single-haplotype resolution and estimate tumor purity and ploidy using long-read sequencing data with or without a germline control sample. We also establish best practices for benchmarking SV detection algorithms across the entire genome in a data-driven manner using replication and read-backed phasing analysis. Through the analysis of matched Illumina and nanopore whole-genome sequencing data for 99 human tumor-normal pairs, we show that SAVANA has significantly higher sensitivity and 13- and 82-times-higher specificity than the second and third-best performing algorithms. Moreover, SVs reported by SAVANA are highly consistent with those detected using short-read sequencing. In summary, SAVANA enables the application of long-read sequencing to detect SVs and SCNAs reliably.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: H.E. and C.M.S. have received travel bursaries from ONT. The other authors declare no competing interests.
Figures



Similar articles
-
Expectations and blind spots for structural variation detection from long-read assemblies and short-read genome sequencing technologies.Am J Hum Genet. 2021 May 6;108(5):919-928. doi: 10.1016/j.ajhg.2021.03.014. Epub 2021 Mar 30. Am J Hum Genet. 2021. PMID: 33789087 Free PMC article.
-
Can a Liquid Biopsy Detect Circulating Tumor DNA With Low-passage Whole-genome Sequencing in Patients With a Sarcoma? A Pilot Evaluation.Clin Orthop Relat Res. 2025 Jan 1;483(1):39-48. doi: 10.1097/CORR.0000000000003161. Epub 2024 Jun 21. Clin Orthop Relat Res. 2025. PMID: 38905450
-
SCOPE: A Normalization and Copy-Number Estimation Method for Single-Cell DNA Sequencing.Cell Syst. 2020 May 20;10(5):445-452.e6. doi: 10.1016/j.cels.2020.03.005. Cell Syst. 2020. PMID: 32437686 Free PMC article.
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
-
Artificial intelligence for diagnosing exudative age-related macular degeneration.Cochrane Database Syst Rev. 2024 Oct 17;10(10):CD015522. doi: 10.1002/14651858.CD015522.pub2. Cochrane Database Syst Rev. 2024. PMID: 39417312
Cited by
-
Comprehensive genomic characterization of early-stage bladder cancer.Nat Genet. 2025 Jan;57(1):115-125. doi: 10.1038/s41588-024-02030-z. Epub 2025 Jan 3. Nat Genet. 2025. PMID: 39753772 Free PMC article.
-
Genome-wide association testing beyond SNPs.Nat Rev Genet. 2025 Mar;26(3):156-170. doi: 10.1038/s41576-024-00778-y. Epub 2024 Oct 7. Nat Rev Genet. 2025. PMID: 39375560 Free PMC article. Review.
References
-
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature578, 82–93 (2020).
-
- Cortés-Ciriano, I., Gulhan, D. C., Lee, J. J.-K., Melloni, G. E. M. & Park, P. J. Computational analysis of cancer genome sequencing data. Nat. Rev. Genet.23, 298–314 (2021). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
