

SAVANA: reliable analysis of somatic structural variants and copy number aberrations using long-read sequencing
- PMID: 40437218
- PMCID: PMC12240814
- DOI: 10.1038/s41592-025-02708-0
SAVANA: reliable analysis of somatic structural variants and copy number aberrations using long-read sequencing
Abstract
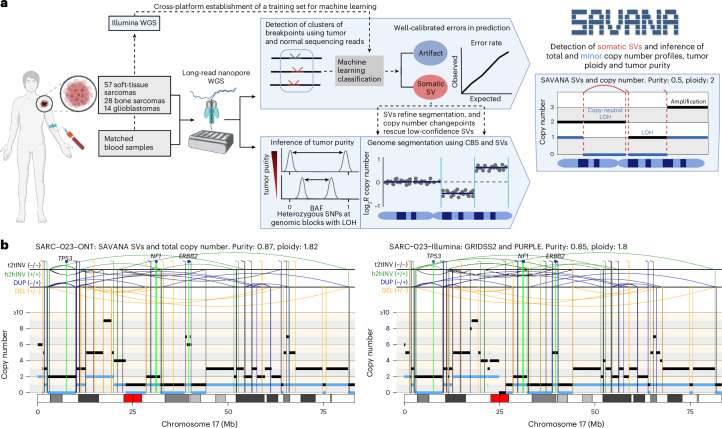
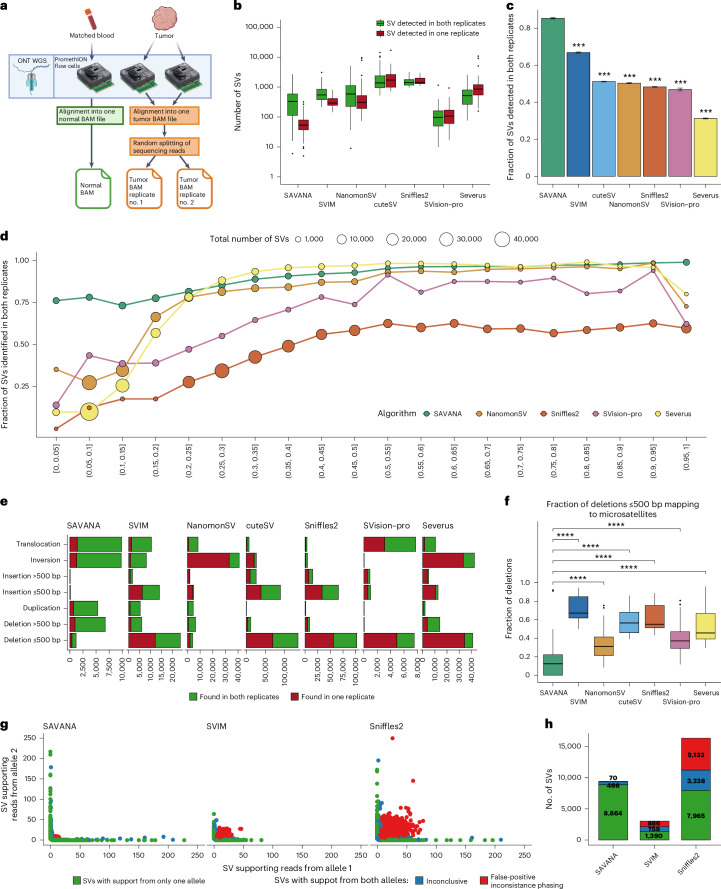
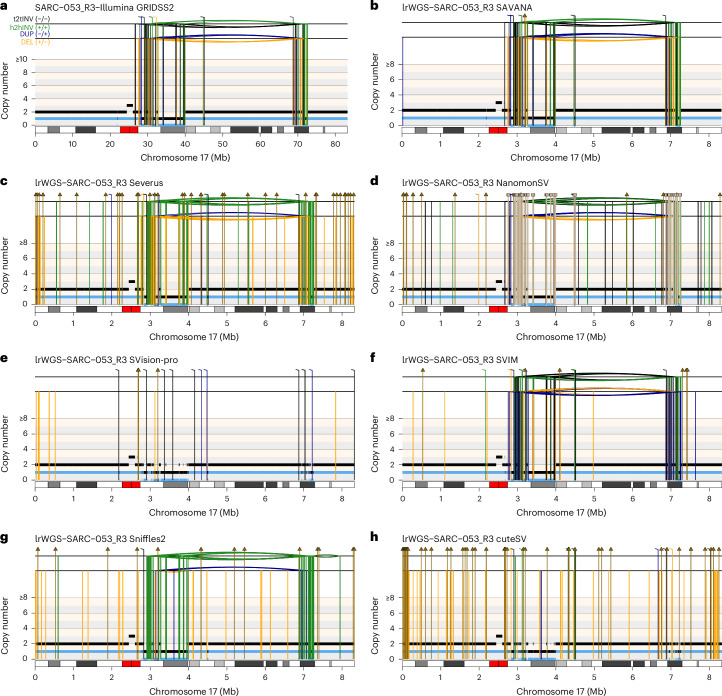
Accurate detection of somatic structural variants (SVs) and somatic copy number aberrations (SCNAs) is critical to study the mutational processes underpinning cancer evolution. Here we describe SAVANA, an algorithm designed to detect somatic SVs and SCNAs at single-haplotype resolution and estimate tumor purity and ploidy using long-read sequencing data with or without a germline control sample. We also establish best practices for benchmarking SV detection algorithms across the entire genome in a data-driven manner using replication and read-backed phasing analysis. Through the analysis of matched Illumina and nanopore whole-genome sequencing data for 99 human tumor-normal pairs, we show that SAVANA has significantly higher sensitivity and 13- and 82-times-higher specificity than the second and third-best performing algorithms. Moreover, SVs reported by SAVANA are highly consistent with those detected using short-read sequencing. In summary, SAVANA enables the application of long-read sequencing to detect SVs and SCNAs reliably.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: H.E. and C.M.S. have received travel bursaries from ONT. The other authors declare no competing interests.
Figures



References
-
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature578, 82–93 (2020).
-
- Cortés-Ciriano, I., Gulhan, D. C., Lee, J. J.-K., Melloni, G. E. M. & Park, P. J. Computational analysis of cancer genome sequencing data. Nat. Rev. Genet.23, 298–314 (2021). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
