

Case report of Lafora disease: a rare genetic disorder manifesting as progressive myoclonic epilepsy
- PMID: 40442642
- PMCID: PMC12121156
- DOI: 10.1186/s12883-025-04253-x
Case report of Lafora disease: a rare genetic disorder manifesting as progressive myoclonic epilepsy
Abstract
Background: Lafora disease (LD) is a rare, autosomal recessive progressive myoclonic epilepsy caused by mutations in EPM2A or EPM2B. It is characterized by abnormal glycogen metabolism leading to poly-glucosan deposits, known as Lafora bodies, in various tissues. LD typically manifests during adolescence with progressive neurological decline, including myoclonic seizures, cognitive impairment, and ataxia. Early diagnosis is critical for symptom management, yet the disease remains challenging to treat due to its refractory nature.
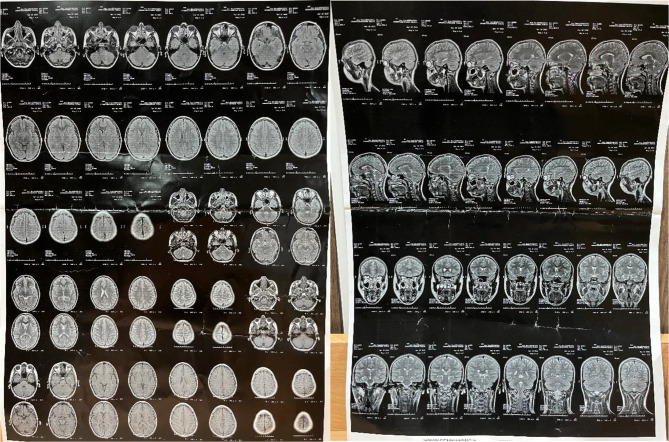
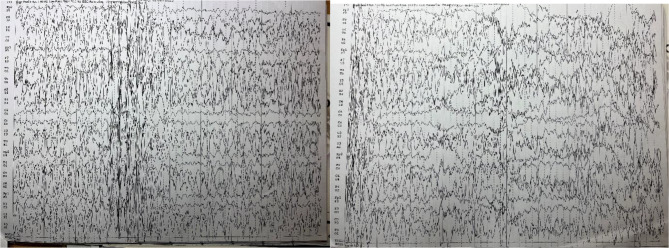
Case presentation: We report the case of a 15-year-old male who initially presented with tonic-clonic and myoclonic seizures, bilateral lower limb paralysis, and hand tremors. Despite normal initial imaging findings, subsequent clinical progression raised suspicion for progressive myoclonic epilepsy. Genetic testing identified a homozygous pathogenic variant in EPM2A, confirming the diagnosis of LD. electroencephalogram (EEG) findings evolved over time, showing generalized spikes, poly-spikes, and spike-wave complexes on a slow background, consistent with advanced LD. The patient's seizures proved refractory to standard anti-epileptic drugs, necessitating the addition of phenobarbital, metformin, and zonisamide, which eventually achieved partial seizure control. Family genetic screening identified heterozygous carriers without clinical symptoms, emphasizing the need for genetic counseling.
Conclusions: This case highlights the diagnostic challenges of LD, particularly in its early stages when clinical and imaging findings may be nonspecific. The report underscores the importance of genetic testing in confirming the diagnosis and tailoring management strategies. Despite limited treatment options, individualized multi-drug regimens may help achieve partial symptom control. Early recognition and comprehensive management, including family counseling, are essential in improving quality of life for patients and their families.
Keywords: EPM2A mutation; Lafora diseas; Neurological decline; Progressive myoclonic epilepsy; Refractory epilepsy.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Written informed consent was obtained from the participants and/or their parents/legal guardians for publication of the case report. Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Novel mutation of EPM2A causes progressive myoclonic epilepsy: a case report.Neurol Sci. 2022 May;43(5):3467-3471. doi: 10.1007/s10072-022-05986-0. Epub 2022 Mar 7. Neurol Sci. 2022. PMID: 35257260
-
A novel compound heterozygous EPM2A mutation in a Chinese boy with Lafora disease.Neurol Sci. 2020 Aug;41(8):2267-2270. doi: 10.1007/s10072-020-04377-7. Epub 2020 Apr 28. Neurol Sci. 2020. PMID: 32342326
-
The presenting symptoms of Lafora Disease: An electroclinical and genetic study in five Apulian (Southern Italy) families.Seizure. 2020 Dec;83:145-153. doi: 10.1016/j.seizure.2020.10.022. Epub 2020 Oct 27. Seizure. 2020. PMID: 33152654
-
Lafora progressive myoclonus epilepsy: recent insights into cell degeneration.Recent Pat Endocr Metab Immune Drug Discov. 2012 May;6(2):99-107. doi: 10.2174/187221412800604617. Recent Pat Endocr Metab Immune Drug Discov. 2012. PMID: 22369717 Review.
-
Lafora disease.Epileptic Disord. 2016 Sep 1;18(S2):38-62. doi: 10.1684/epd.2016.0842. Epileptic Disord. 2016. PMID: 27702709 Free PMC article. Review.
References
-
- Malek N, Stewart W, Greene J. The progressive myoclonic epilepsies. Pract Neurol. 2015;15(3):164–71. - PubMed
-
- Andrade DM, Ackerley CA, Minett TSC, Teive HAG, Bohlega S, Scherer SW, et al. Skin biopsy in Lafora disease: genotype–phenotype correlations and diagnostic pitfalls. Neurology. 2003;61(11):1611–4. - PubMed
-
- Barbieri F, Santangelo R, Gasparo-Rippa P, Santoro M. Biopsy findings (cerebral cortex, muscle, skin) in Lafora disease. Acta Neurol. 1987;9(2):81–94. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
