

Simvastatin overcomes the pPCK1-pLDHA-SPRINGlac axis-mediated ferroptosis and chemo-immunotherapy resistance in AKT-hyperactivated intrahepatic cholangiocarcinoma
- PMID: 40443016
- PMCID: PMC12365544
- DOI: 10.1002/cac2.70036
Simvastatin overcomes the pPCK1-pLDHA-SPRINGlac axis-mediated ferroptosis and chemo-immunotherapy resistance in AKT-hyperactivated intrahepatic cholangiocarcinoma
Abstract
Background: Intrahepatic cholangiocarcinoma (ICC) is a challenging cancer with an increasing incidence. The Phase III TOPAZ-1/KEYNOTE-966 study demonstrated chemo-immunotherapy (CIT) as a significant advancement, potentially replacing traditional chemotherapy for advanced biliary tract cancer. Ferroptosis is a crucial process that affects cancer cell survival and therapy resistance. Although AKT hyperactivation is prevalent in numerous cancers, including ICC, its role in ferroptosis resistance remains unclear. This study explored whether targeting ferroptosis can enhance CIT response rates, specifically in ICC patients with AKT hyperactivation.
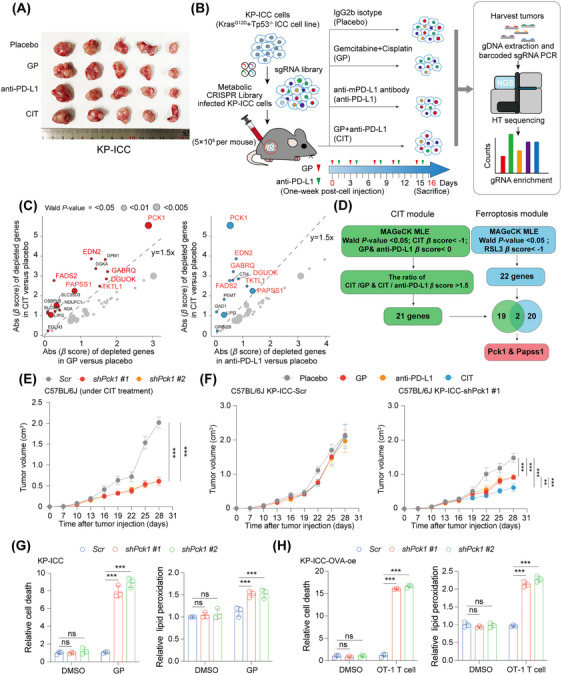
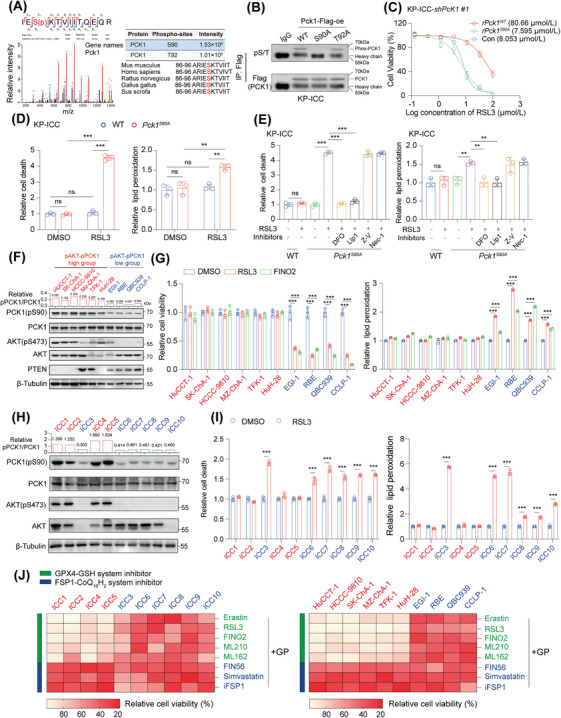
Methods: In vivo metabolic CRISPR screening in a KrasG12D/Tp53-/- ICC mouse model was used to identify primary regulators of ferroptosis during CIT (gemcitabine, cisplatin, and anti-mouse programmed cell death 1 ligand 1). Phosphoenolpyruvate carboxykinase 1 (PCK1) was assessed for its role in ferroptosis and treatment resistance in preclinical models under AKT activation levels. Molecular and biochemical techniques were used to explore PCK1-related resistance mechanisms in AKT-hyperactivated ICC.
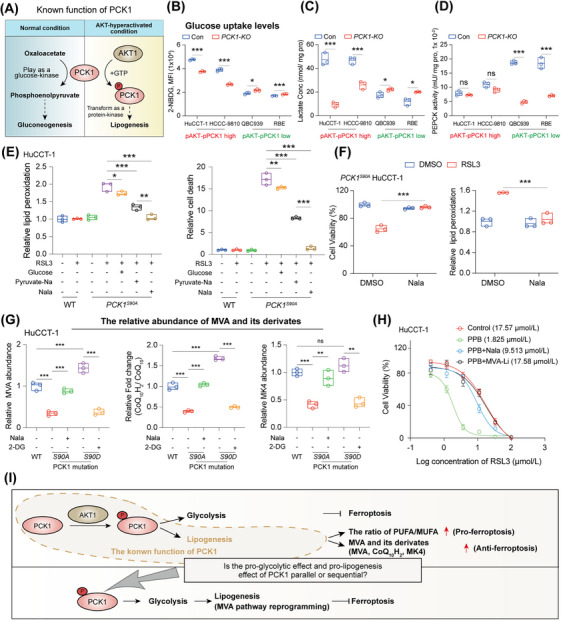
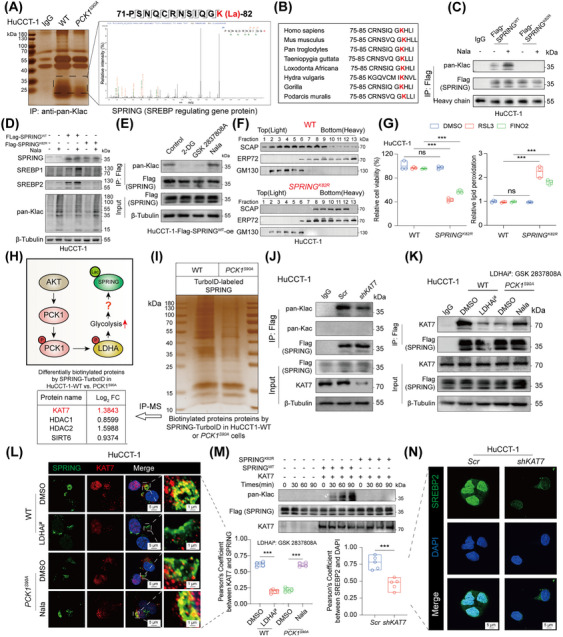
Results: Under AKT hyperactivation condition, phosphorylated PCK1 (pPCK1) promoted metabolic reprogramming, enhancing ubiquinol and menaquinone-4 synthesis through the mevalonate (MVA) pathway. This cascade was mediated by the pPCK1-pLDHA-SPRINGlac axis. Inhibiting PCK1 phosphorylation or using simvastatin significantly augmented CIT efficacy in preclinical models. Clinical data further indicated that phosphorylated AKT (pAKT)-pPCK1 levels might serve as a biomarker to predict CIT response in ICC.
Conclusion: This study identified the pAKT-pPCK1-pLDHA-SPRINGlac axis as a novel mechanism driving ferroptosis resistance in AKT-hyperactivated ICC by associating glycolytic activation with MVA flux reprogramming. Targeting this axis, potentially through statin-based therapies, may offer a strategy to sensitize ICC cells to ferroptosis and improve treatment outcomes.
Keywords: Chemo‐immunotherapy; Ferroptosis; Intrahepatic cholangiocarcinoma; Lactylation; Mevalonate pathway; PCK1.
© 2025 The Author(s). Cancer Communications published by John Wiley & Sons Australia, Ltd. on behalf of Sun Yat‐sen University Cancer Center.
Conflict of interest statement
The authors declare no potential conflicts of interest.
Figures




References
-
- Oh D‐Y, Ruth He A, Qin S, Chen L‐T, Okusaka T, Vogel A, et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer. NEJM Evid. 2022;1(8):EVIDoa2200015. - PubMed
-
- Satriano L, Lewinska M, Rodrigues PM, Banales JM, Andersen JB. Metabolic rearrangements in primary liver cancers: cause and consequences. Nat Rev Gastroenterol Hepatol. 2019;16(12):748‐66. - PubMed
MeSH terms
Substances
Grants and funding
- 81874149/National Natural Science Foundation of China
- 82171834/National Natural Science Foundation of China
- 82303832/National Natural Science Foundation of China
- 82403618/National Natural Science Foundation of China
- 2024M761039/China Postdoctoral Science Foundation
- BX20240129/China National Postdoctoral Program for Innovative Talents
- 2024HBBHCXA035/Postdoctor Project of Hubei Province
- 2022020801020456/Knowledge Innovation Program of Wuhan-Shuguang Project
- BE2022725/Key Research and Development Program of Social Development of Jiangsu Province
- SKY2023049/Suzhou Science and Technology Development Plan Project
- Jiangsu Province Seventh 333 High Level (Second Level) Talents Project
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
