

A robust multiplex-DIA workflow profiles protein turnover regulations associated with cisplatin resistance and aneuploidy
- PMID: 40447611
- PMCID: PMC12125295
- DOI: 10.1038/s41467-025-60319-x
A robust multiplex-DIA workflow profiles protein turnover regulations associated with cisplatin resistance and aneuploidy
Abstract
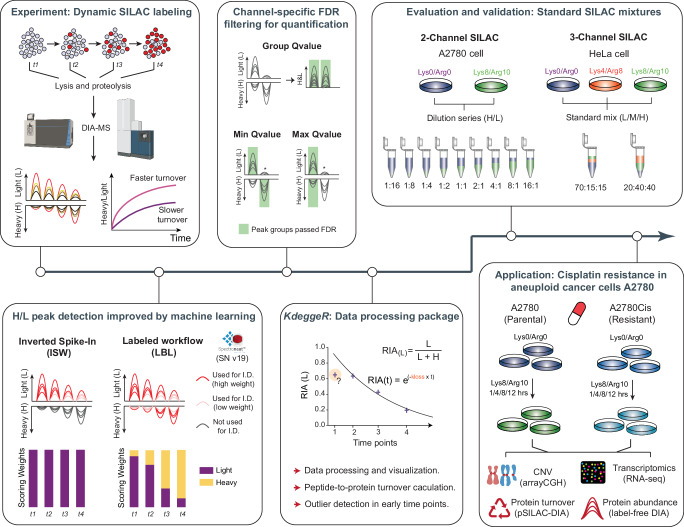
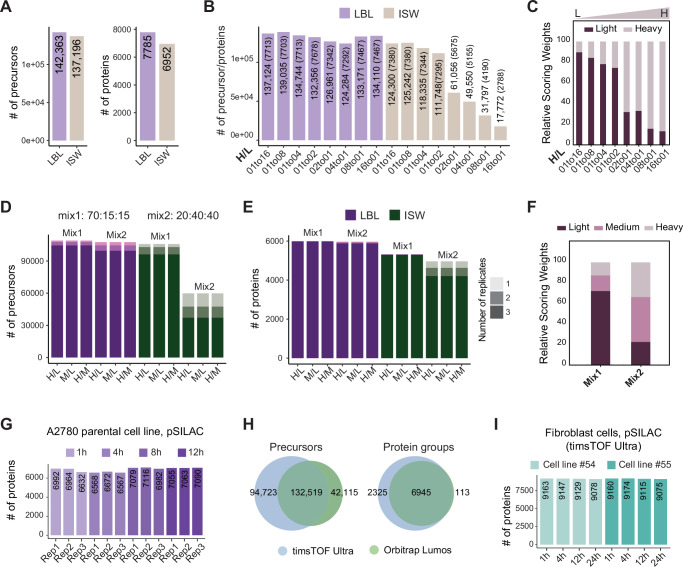
Quantifying protein turnover is fundamental to understanding cellular processes and advancing drug discovery. Multiplex-DIA mass spectrometry (MS), combined with dynamic SILAC labeling (pulse-SILAC, or pSILAC) reliably measures protein turnover and degradation kinetics. Previous multiplex-DIA-MS workflows have employed various strategies including leveraging the highest isotopic labeling channels to enhance the detection of isotopic signal pairs. Here we present a robust workflow that integrates a machine learning algorithm and channel-specific statistical filtering, enabling dynamic adaptation to channel ratio changes across multiplexed experiments and enhancing both coverage and accuracy of protein turnover profiling. We also introduce KdeggeR, a data analysis tool optimized for pSILAC-DIA experiments, which determines and visualizes peptide and protein degradation profiles. Our workflow is broadly applicable, as demonstrated on 2-channel and 3-channel DIA datasets and across two MS platforms. Applying this framework to an aneuploid cancer cell model before and after cisplatin resistance, we uncover strong proteome buffering of key protein complex subunits encoded by the aneuploid genome mediated by protein degradation. We identify resistance-associated turnover signatures, including mitochondrial metabolic adaptation via accelerated degradation of respiratory complexes I and IV. Our approach provides a powerful platform for high-throughput, quantitative analysis of proteome dynamics and stability in health and disease.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: O.B., T.G. and L.R. are employees of Biognosys AG. Spectronaut is a trademark of Biognosys AG. The remaining authors declare no competing interests.
Figures




Update of
-
A Comprehensive and Robust Multiplex-DIA Workflow Profiles Protein Turnover Regulations Associated with Cisplatin Resistance.bioRxiv [Preprint]. 2024 Oct 31:2024.10.28.620709. doi: 10.1101/2024.10.28.620709. bioRxiv. 2024. Update in: Nat Commun. 2025 May 30;16(1):5034. doi: 10.1038/s41467-025-60319-x. PMID: 39554001 Free PMC article. Updated. Preprint.
Similar articles
-
A Comprehensive and Robust Multiplex-DIA Workflow Profiles Protein Turnover Regulations Associated with Cisplatin Resistance.bioRxiv [Preprint]. 2024 Oct 31:2024.10.28.620709. doi: 10.1101/2024.10.28.620709. bioRxiv. 2024. Update in: Nat Commun. 2025 May 30;16(1):5034. doi: 10.1038/s41467-025-60319-x. PMID: 39554001 Free PMC article. Updated. Preprint.
-
BoxCarmax: A High-Selectivity Data-Independent Acquisition Mass Spectrometry Method for the Analysis of Protein Turnover and Complex Samples.Anal Chem. 2021 Feb 16;93(6):3103-3111. doi: 10.1021/acs.analchem.0c04293. Epub 2021 Feb 3. Anal Chem. 2021. PMID: 33533601 Free PMC article.
-
Toward a hypothesis-free understanding of how phosphorylation dynamically impacts protein turnover.Proteomics. 2023 Feb;23(3-4):e2100387. doi: 10.1002/pmic.202100387. Epub 2022 Dec 7. Proteomics. 2023. PMID: 36422574 Free PMC article.
-
Stable isotope labeling by amino acids in cell culture (SILAC) for quantitative proteomics.Adv Exp Med Biol. 2014;806:93-106. doi: 10.1007/978-3-319-06068-2_5. Adv Exp Med Biol. 2014. PMID: 24952180 Review.
-
Standardization approaches in absolute quantitative proteomics with mass spectrometry.Mass Spectrom Rev. 2018 Nov;37(6):715-737. doi: 10.1002/mas.21542. Epub 2017 Jul 31. Mass Spectrom Rev. 2018. PMID: 28758227 Review.
References
-
- Venable, J. D., Dong, M. Q., Wohlschlegel, J., Dillin, A. & Yates, J. R. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods1, 39–45 (2004). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
