

DeepDTAGen: a multitask deep learning framework for drug-target affinity prediction and target-aware drugs generation
- PMID: 40447614
- PMCID: PMC12125237
- DOI: 10.1038/s41467-025-59917-6
DeepDTAGen: a multitask deep learning framework for drug-target affinity prediction and target-aware drugs generation
Abstract
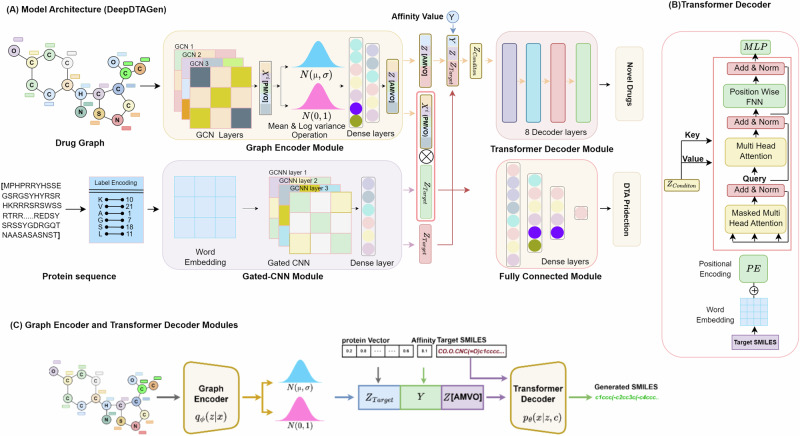
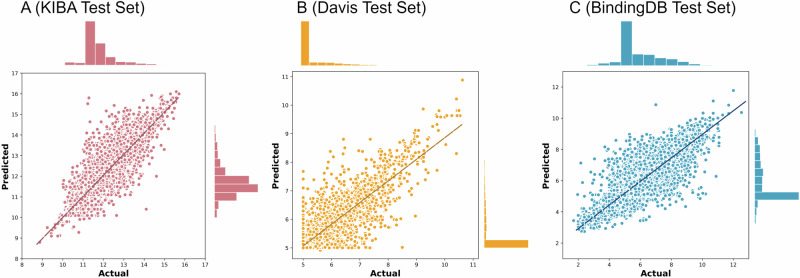
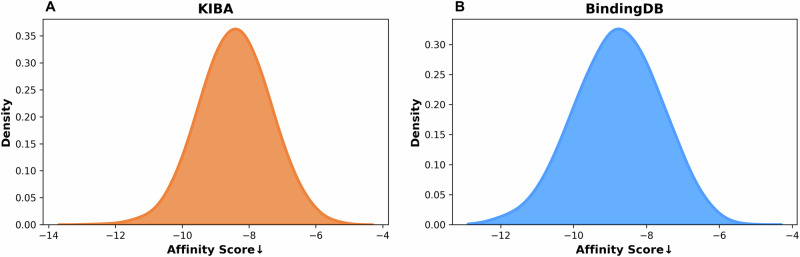
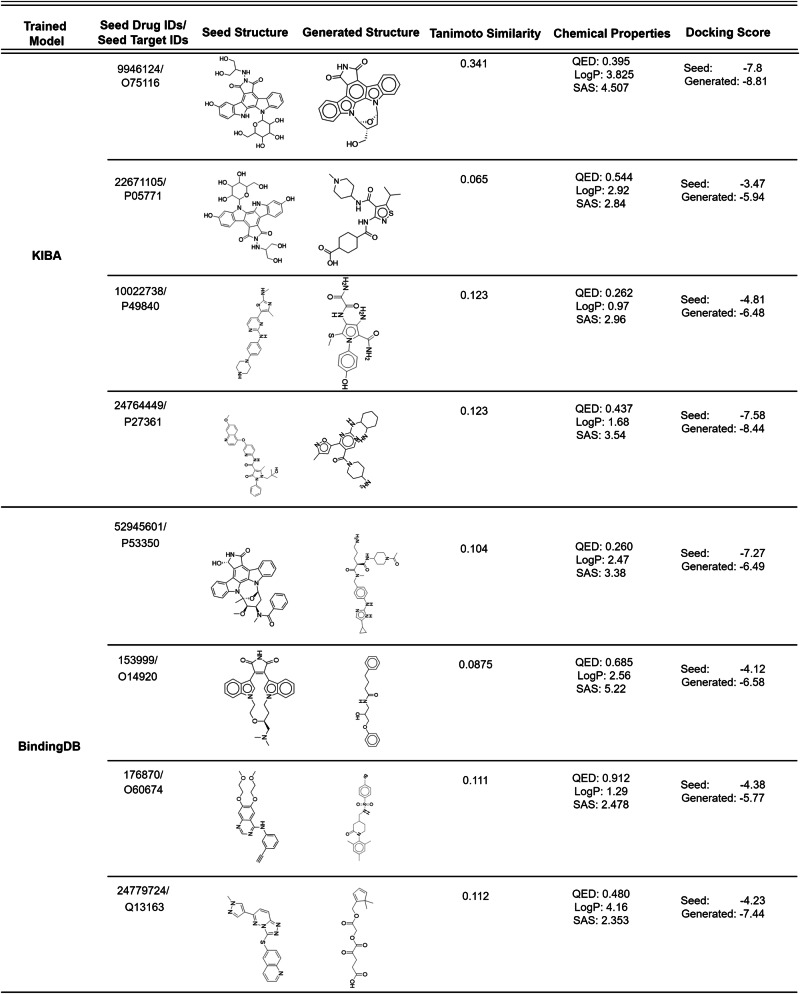
Identifying novel drugs that can interact with target proteins is a highly challenging, time-consuming, and costly task in drug discovery and development. Numerous machine learning-based models have recently been utilized to accelerate the drug discovery process. However, these existing methods are primarily uni-tasking, either designed to predict drug-target interaction (DTI) or generate new drugs. Through the lens of pharmacological research, these tasks are intrinsically interconnected and play a critical role in effective drug development. Therefore, the learning models must be utilized in such a manner to learn the structural properties of drug molecules, the conformational dynamics of proteins, and the bioactivity between drugs and targets. To this end, this paper develops a novel multitask learning framework that can predict drug-target binding affinities and simultaneously generate new target-aware drug variants, using common features for both tasks. In addition, we developed the FetterGrad algorithm to address the optimization challenges associated with multitask learning particularly those caused by gradient conflicts between distinct tasks. Comprehensive experiments on three real-world datasets demonstrate that the proposed model provides an effective mechanism for predicting drug-target binding affinities and generating novel drugs, thus greatly facilitating the drug discovery process.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
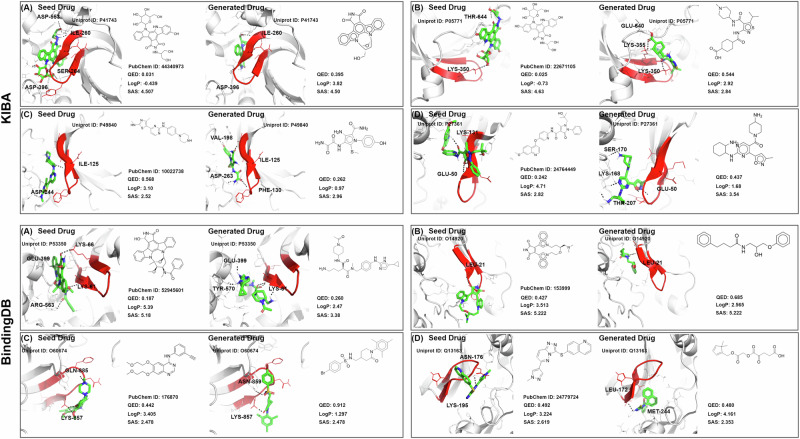
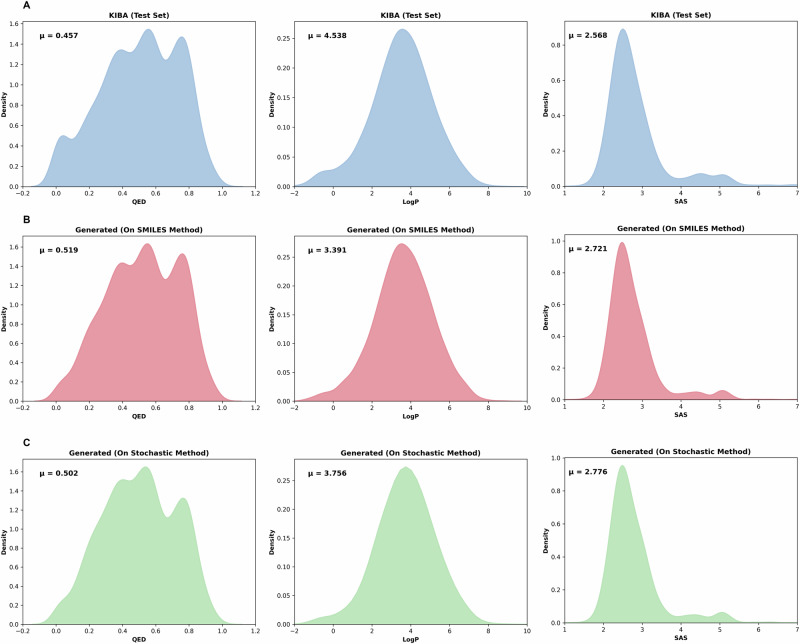
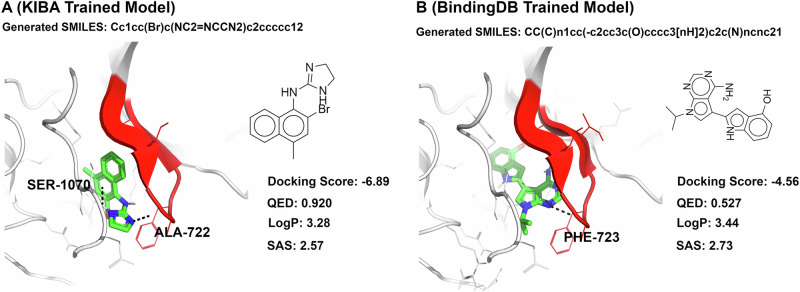
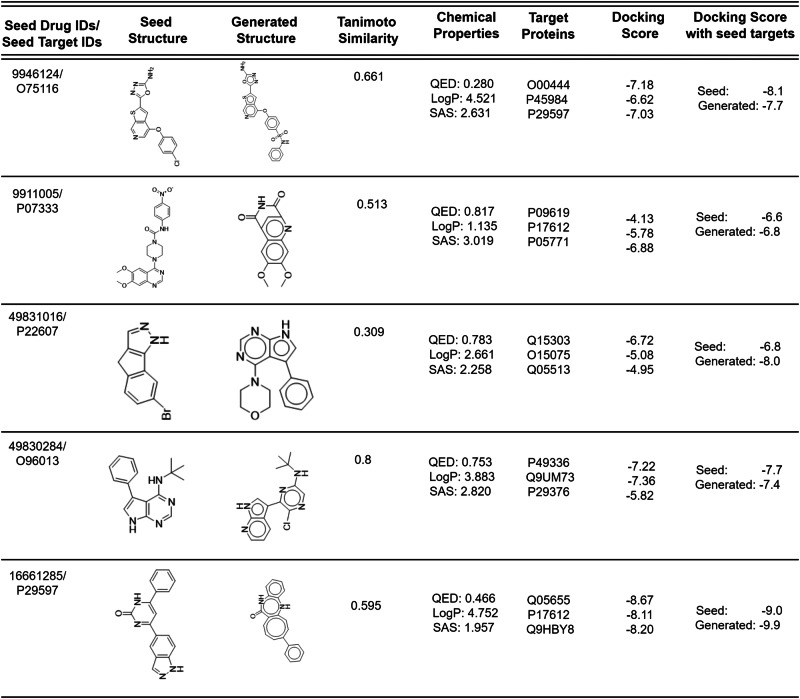
Similar articles
-
CGPDTA: An Explainable Transfer Learning-Based Predictor With Molecule Substructure Graph for Drug-Target Binding Affinity.J Comput Chem. 2025 Jan 5;46(1):e27538. doi: 10.1002/jcc.27538. J Comput Chem. 2025. PMID: 39653581
-
TC-DTA: Predicting Drug-Target Binding Affinity With Transformer and Convolutional Neural Networks.IEEE Trans Nanobioscience. 2024 Oct;23(4):572-578. doi: 10.1109/TNB.2024.3441590. Epub 2024 Oct 15. IEEE Trans Nanobioscience. 2024. PMID: 39133595
-
Drug-target interaction prediction using Multi Graph Regularized Nuclear Norm Minimization.PLoS One. 2020 Jan 16;15(1):e0226484. doi: 10.1371/journal.pone.0226484. eCollection 2020. PLoS One. 2020. PMID: 31945078 Free PMC article.
-
Machine learning models for drug-target interactions: current knowledge and future directions.Drug Discov Today. 2020 Apr;25(4):748-756. doi: 10.1016/j.drudis.2020.03.003. Epub 2020 Mar 12. Drug Discov Today. 2020. PMID: 32171918 Review.
-
Artificial intelligence to deep learning: machine intelligence approach for drug discovery.Mol Divers. 2021 Aug;25(3):1315-1360. doi: 10.1007/s11030-021-10217-3. Epub 2021 Apr 12. Mol Divers. 2021. PMID: 33844136 Free PMC article. Review.
Cited by
-
AI-Driven Polypharmacology in Small-Molecule Drug Discovery.Int J Mol Sci. 2025 Jul 21;26(14):6996. doi: 10.3390/ijms26146996. Int J Mol Sci. 2025. PMID: 40725243 Free PMC article. Review.
References
-
- Noble, M. E., Endicott, J. A. & Johnson, L. N. Protein kinase inhibitors: insights into drug design from structure. Science303, 1800–1805 (2004). - PubMed
-
- Wang, K., Zhou, R., Li, Y. & Li, M. DeepDTAF: a deep learning method to predict protein–ligand binding affinity. Brief. Bioinform.22, 072 (2021). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
