

CorrAdjust unveils biologically relevant transcriptomic correlations by efficiently eliminating hidden confounders
- PMID: 40448503
- PMCID: PMC12125544
- DOI: 10.1093/nar/gkaf444
CorrAdjust unveils biologically relevant transcriptomic correlations by efficiently eliminating hidden confounders
Abstract
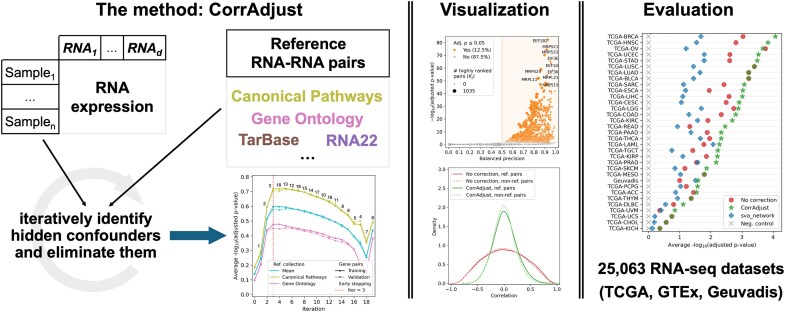
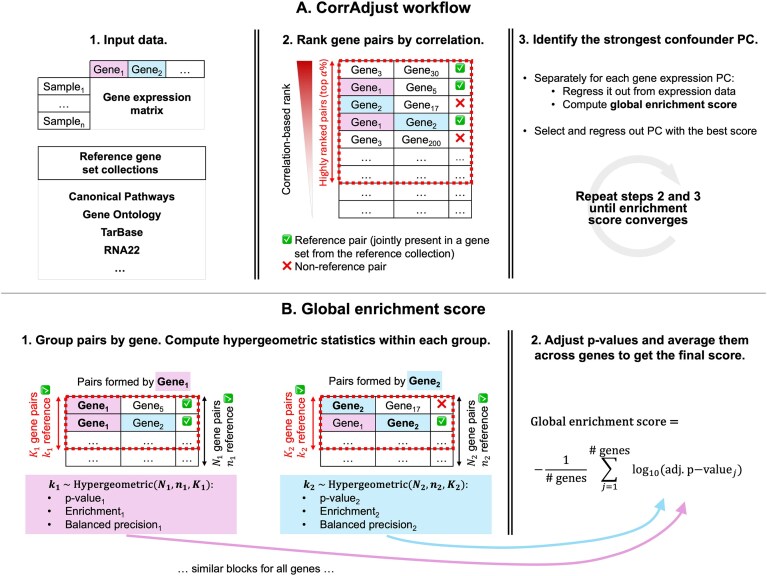
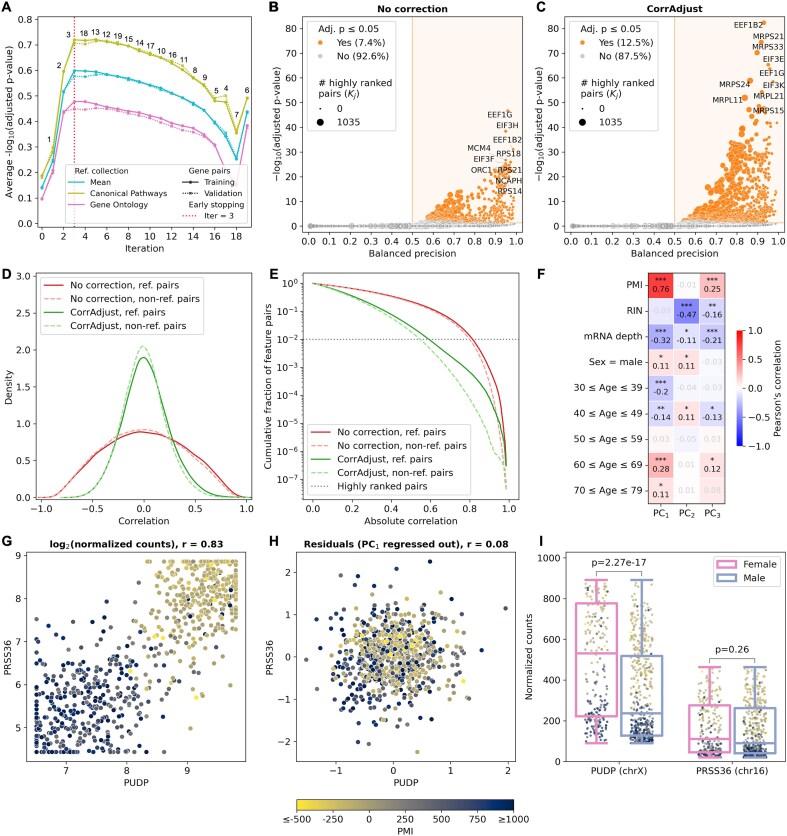
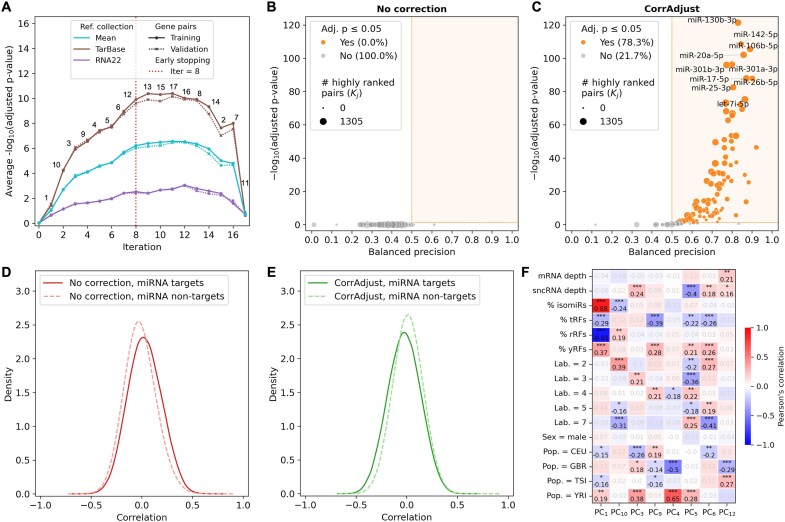
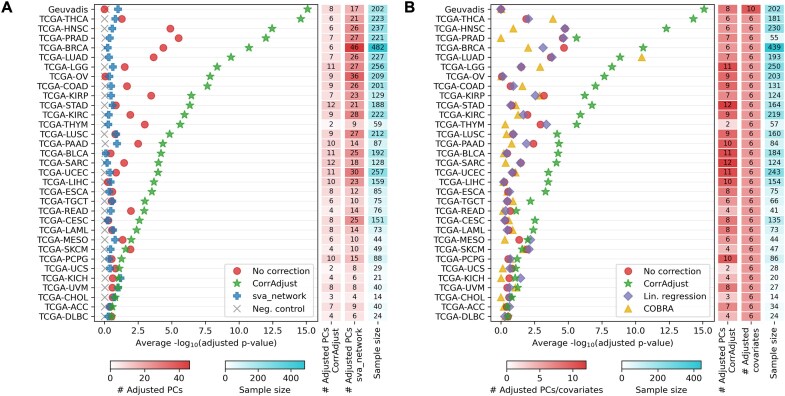
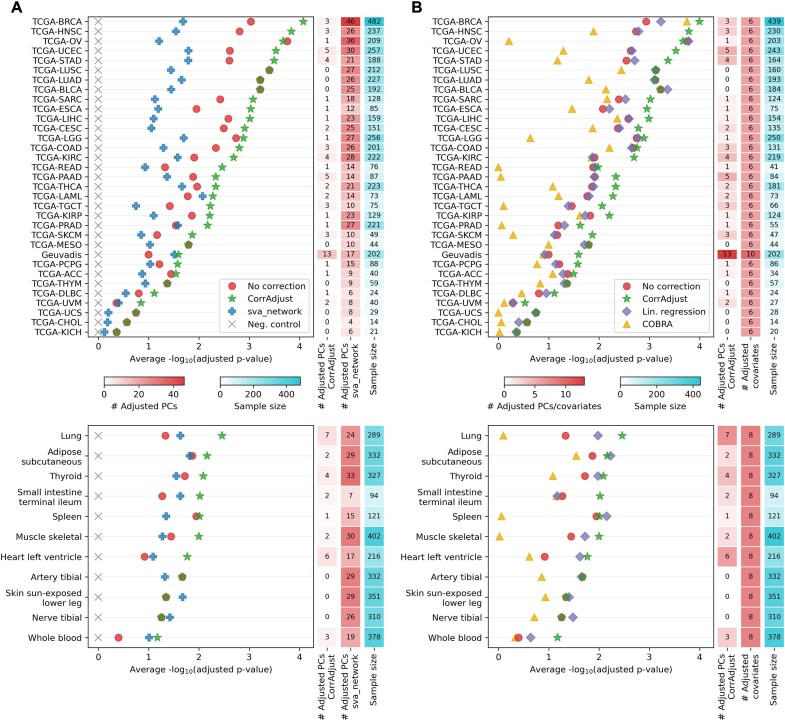
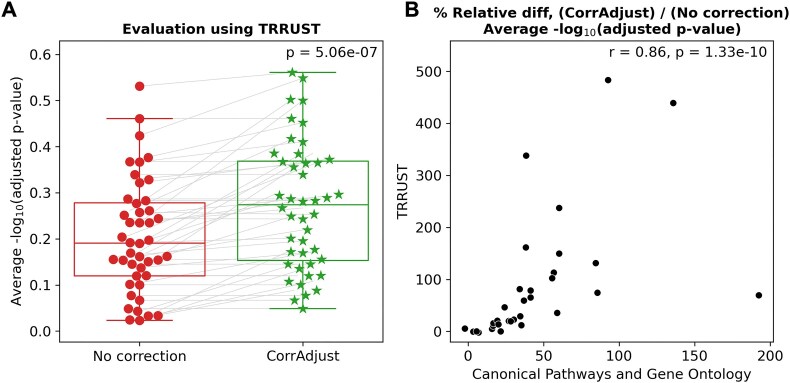
Correcting for confounding variables is often overlooked when computing RNA-RNA correlations, even though it can profoundly affect results. We introduce CorrAdjust, a method for identifying and correcting such hidden confounders. CorrAdjust selects a subset of principal components to residualize from expression data by maximizing the enrichment of "reference pairs" among highly correlated RNA-RNA pairs. Unlike traditional machine learning metrics, this novel enrichment-based metric is specifically designed to evaluate correlation data and provides valuable RNA-level interpretability. CorrAdjust outperforms current state-of-the-art methods when evaluated on 25 063 human RNA-seq datasets from The Cancer Genome Atlas, the Genotype-Tissue Expression project, and the Geuvadis collection. In particular, CorrAdjust excels at integrating small RNA and mRNA sequencing data, significantly enhancing the enrichment of experimentally validated miRNA targets among negatively correlated miRNA-mRNA pairs. CorrAdjust, with accompanying documentation and tutorials, is available at https://tju-cmc-org.github.io/CorrAdjust.
© The Author(s) 2025. Published by Oxford University Press on behalf of Nucleic Acids Research.
Conflict of interest statement
The authors declare that there are no conflicts of interests to disclose.
Figures
Similar articles
-
COME: contrastive mapping learning for spatial reconstruction of single-cell RNA sequencing data.Bioinformatics. 2025 Mar 4;41(3):btaf083. doi: 10.1093/bioinformatics/btaf083. Bioinformatics. 2025. PMID: 39992219 Free PMC article.
-
A Distributed Classifier for MicroRNA Target Prediction with Validation Through TCGA Expression Data.IEEE/ACM Trans Comput Biol Bioinform. 2018 Jul-Aug;15(4):1037-1051. doi: 10.1109/TCBB.2018.2828305. Epub 2018 Apr 19. IEEE/ACM Trans Comput Biol Bioinform. 2018. PMID: 29993641 Free PMC article.
-
Facilitating pathway and network based analysis of RNA-Seq data with pathlinkR.PLoS Comput Biol. 2024 Sep 16;20(9):e1012422. doi: 10.1371/journal.pcbi.1012422. eCollection 2024 Sep. PLoS Comput Biol. 2024. PMID: 39283925 Free PMC article.
-
Characterizing and annotating the genome using RNA-seq data.Sci China Life Sci. 2017 Feb;60(2):116-125. doi: 10.1007/s11427-015-0349-4. Epub 2016 Jun 13. Sci China Life Sci. 2017. PMID: 27294835 Review.
-
Differential Expression Analysis of RNA-seq Reads: Overview, Taxonomy, and Tools.IEEE/ACM Trans Comput Biol Bioinform. 2020 Mar-Apr;17(2):566-586. doi: 10.1109/TCBB.2018.2873010. Epub 2018 Oct 1. IEEE/ACM Trans Comput Biol Bioinform. 2020. PMID: 30281477 Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
