

Transcriptomic analysis of the liver, jejunum, and uterus in different production stages of laying hens
- PMID: 40449105
- PMCID: PMC12164196
- DOI: 10.1016/j.psj.2025.105329
Transcriptomic analysis of the liver, jejunum, and uterus in different production stages of laying hens
Abstract
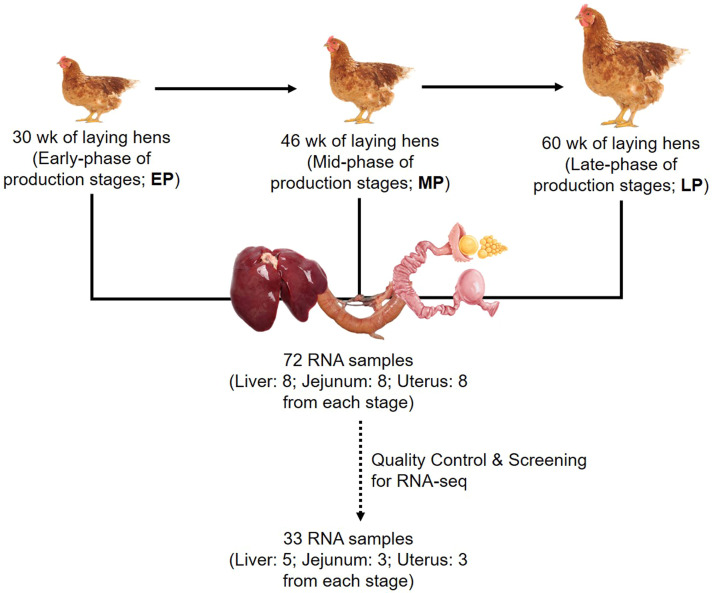
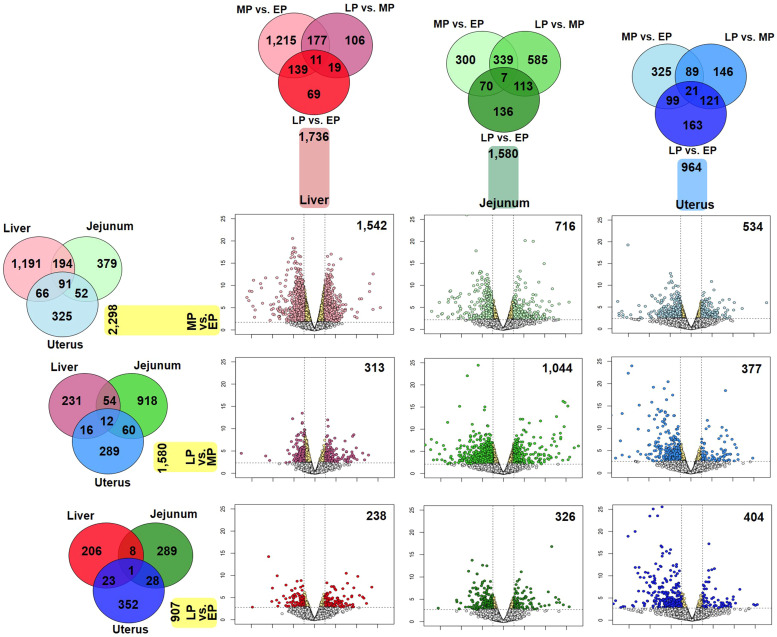
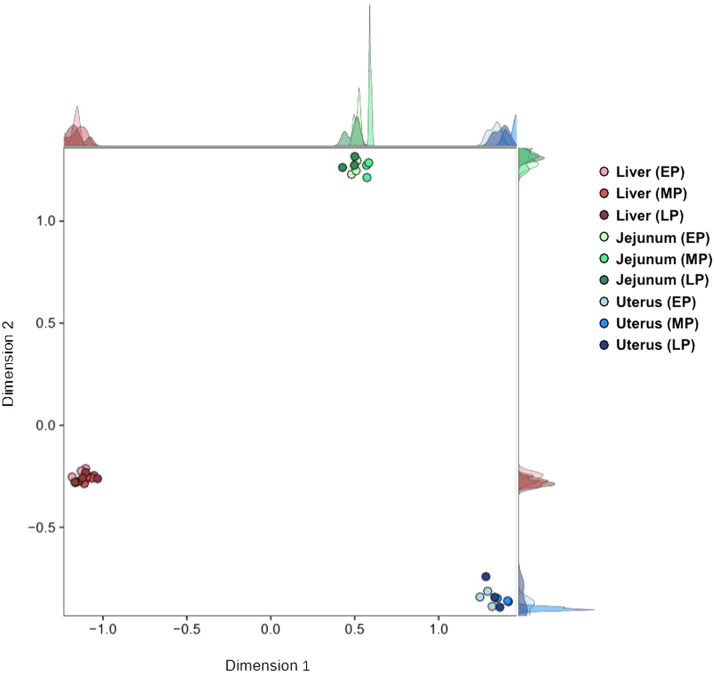
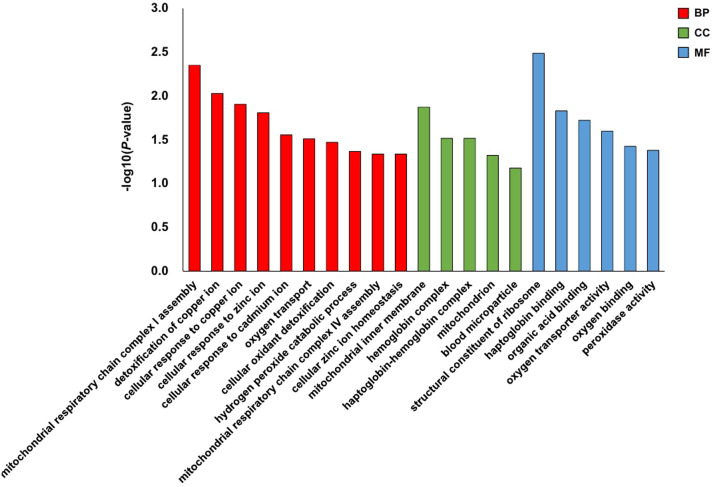
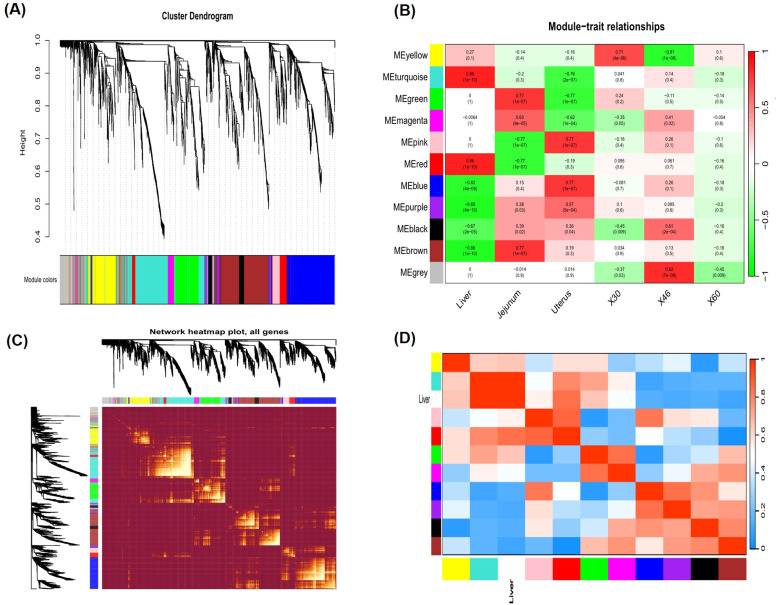
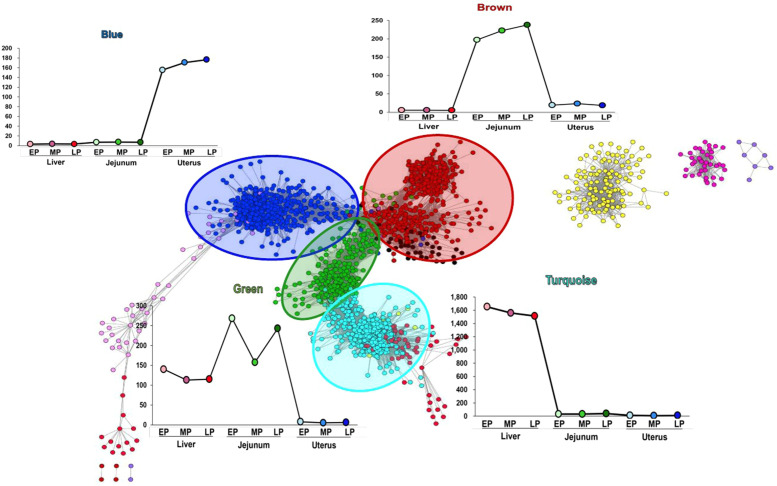
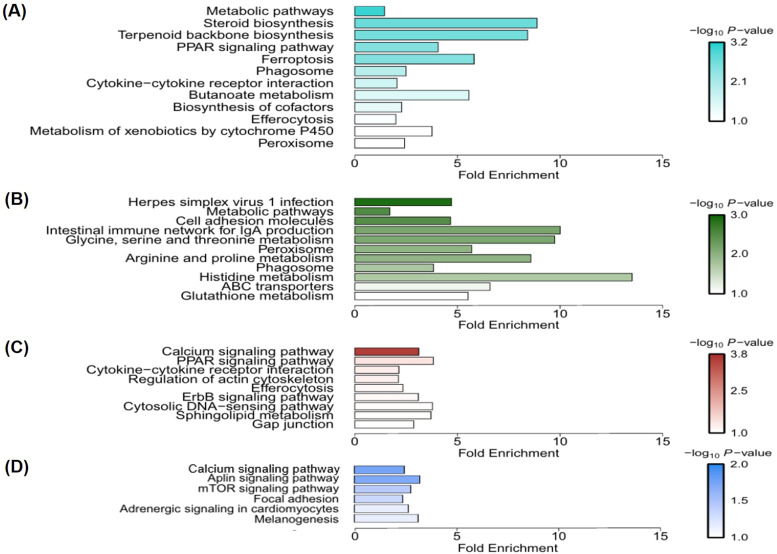
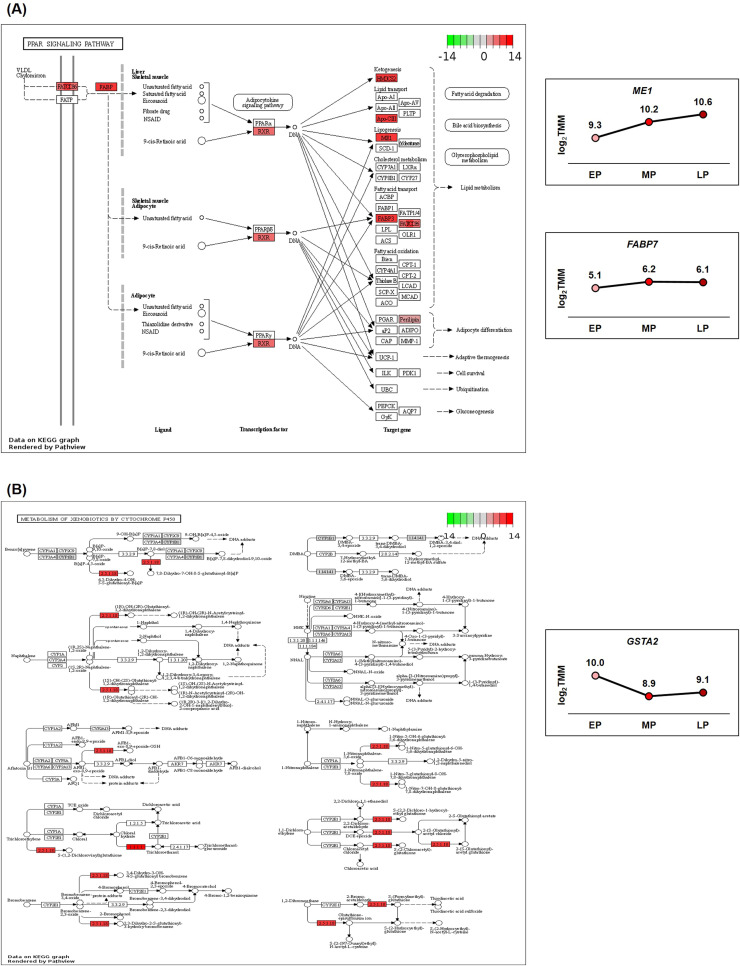
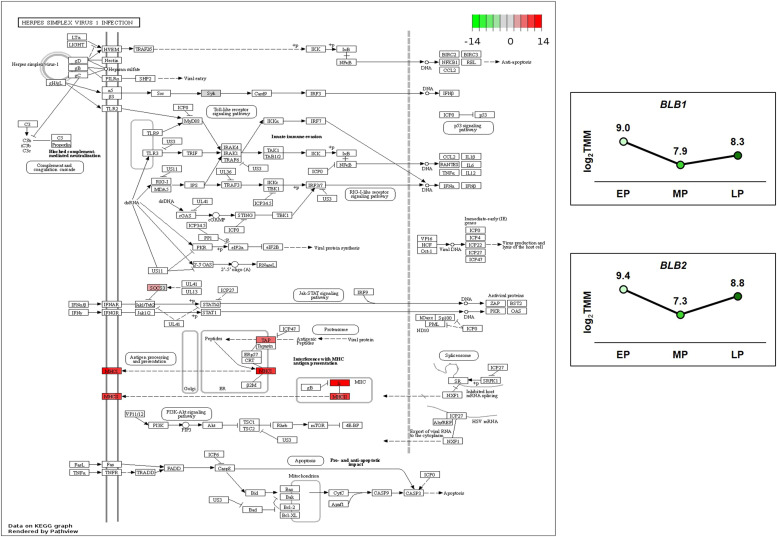
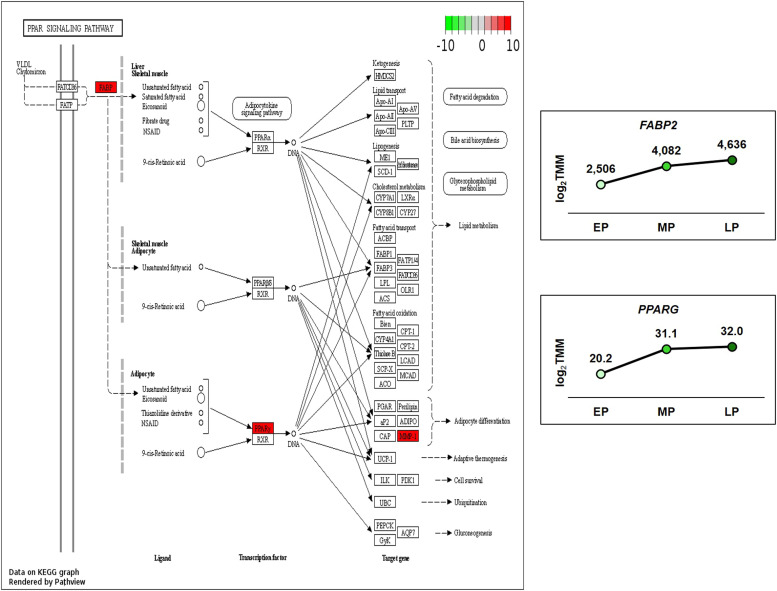
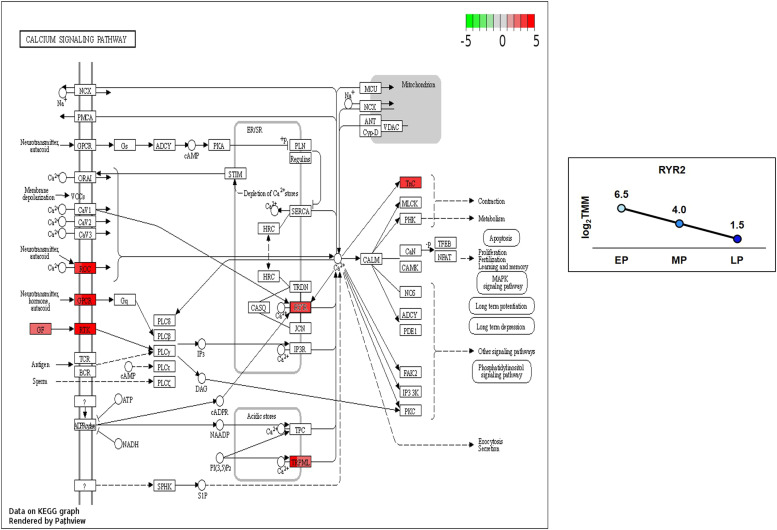
Egg production in laying hens is related to very complex and elaborate processes involving the cooperation of various tissues. Laying hens undergo this complicated production process in different production stages during overall laying periods. However, many previous studies have focused on a single tissue or specific production stage. Thus, we compared multi-tissue transcriptome profiles across different production stages using RNA-seq to understand which overall metabolic changes occur in laying hens as the stage progresses. Laying hens at three distinct production stages of early-phase (EP, 30 wk of age), mid-phase (MP, 46 wk of age), and late-phase (LP, 60 wk of age) were used to analyze transcriptomic changes for the liver, jejunum, and uterus tissues. Weighted gene co-expression network analysis was adopted to detect core modules and central genes, and finally identified 11 co-expression modules. In the liver and jejunum, the expression of genes (e.g., FABP2, FABP7, PPARG) related to fatty acid synthesis was increased with production stages. However, the expression of genes (e.g., GSTA2, BLB1 and BLB2) related to immune responses, including xenobiotic metabolism pathway and the herpes simplex virus 1 infection pathway, was increased in EP compared with other stages. Moreover, the expression of genes related to calcium signaling pathways (e.g., CACNA2D1) and muscle contraction metabolism (e.g., ACTG2 and RYR2) in the uterus was decreased as laying hens were aged. The current findings pave the way for future investigations into the physiological changes in laying hens across different production stages. This research also provides a foundation for elucidating the multi-tissue transcriptome in laying hens and identifying potential genes regulating various biological processes during overall laying periods.
Keywords: Gene co-expression network; Laying hen; Laying stage; Multi-tissue; Transcriptome.
Copyright © 2025. Published by Elsevier Inc.
Conflict of interest statement
Declaration of competing interest The authors declare no conflict of interest for the data presented in this experiment.
Figures
Similar articles
-
Multi-omics analysis reveals associations between host gene expression, gut microbiota, and metabolites in chickens.J Anim Sci. 2024 Jan 3;102:skae263. doi: 10.1093/jas/skae263. J Anim Sci. 2024. PMID: 39243135
-
The promotion of liver vitamin metabolism is of great significance for laying hens during fasting.BMC Genomics. 2025 Jul 1;26(1):603. doi: 10.1186/s12864-025-11730-7. BMC Genomics. 2025. PMID: 40597604 Free PMC article.
-
Eggshell depigmentation in the late phase of production is associated with altered Microbiota and Metabolism of the uterus in laying hens.Poult Sci. 2025 Aug;104(8):105258. doi: 10.1016/j.psj.2025.105258. Epub 2025 May 2. Poult Sci. 2025. PMID: 40367565 Free PMC article.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2021 Apr 19;4(4):CD011535. doi: 10.1002/14651858.CD011535.pub4. Cochrane Database Syst Rev. 2021. Update in: Cochrane Database Syst Rev. 2022 May 23;5:CD011535. doi: 10.1002/14651858.CD011535.pub5. PMID: 33871055 Free PMC article. Updated.
-
A systematic review of potential productivity, egg quality, and animal welfare implications of extended lay cycles in commercial laying hens in Canada.Poult Sci. 2024 Apr;103(4):103475. doi: 10.1016/j.psj.2024.103475. Epub 2024 Feb 1. Poult Sci. 2024. PMID: 38364604 Free PMC article.
References
-
- Adams K.A., Davis A.J. Dietary protein concentration regulates the mRNA expression of chicken hepatic malic enzyme. J. Nutr. 2001;131:2269–2274. - PubMed
-
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
-
- Baltaci A.K., Yuce K., Mogulkoc R. Zinc metabolism and metallothioneins. Biol. Trace Elem. Res. 2018;183:22–31. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
