

varVAMP: degenerate primer design for tiled full genome sequencing and qPCR
- PMID: 40449995
- PMCID: PMC12126543
- DOI: 10.1038/s41467-025-60175-9
varVAMP: degenerate primer design for tiled full genome sequencing and qPCR
Abstract
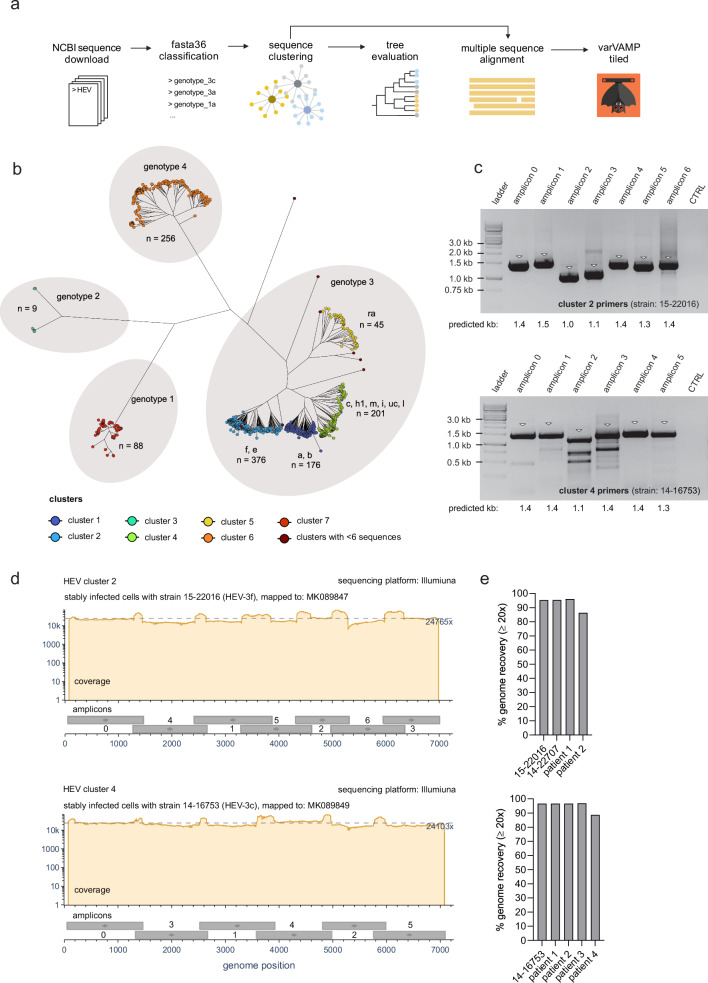
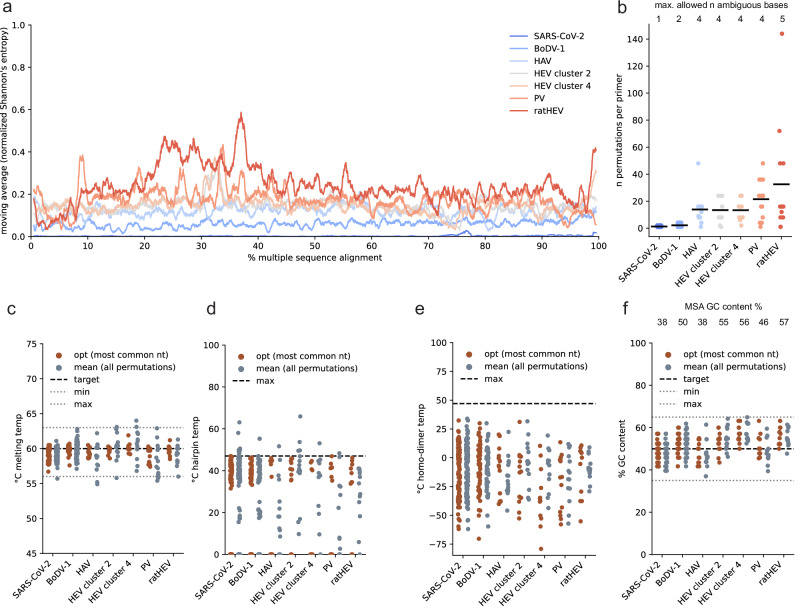
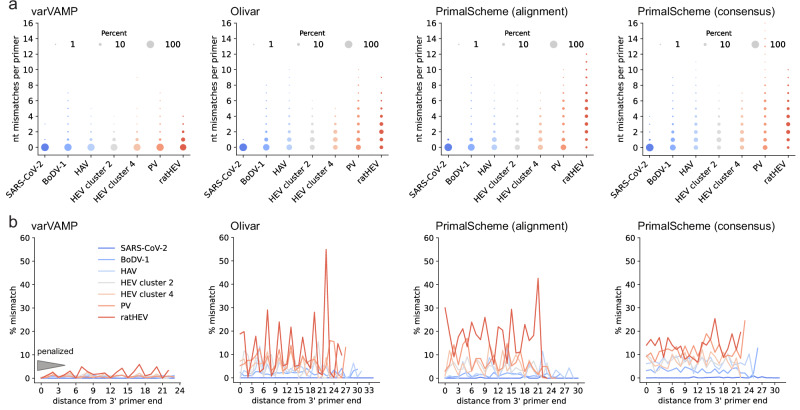
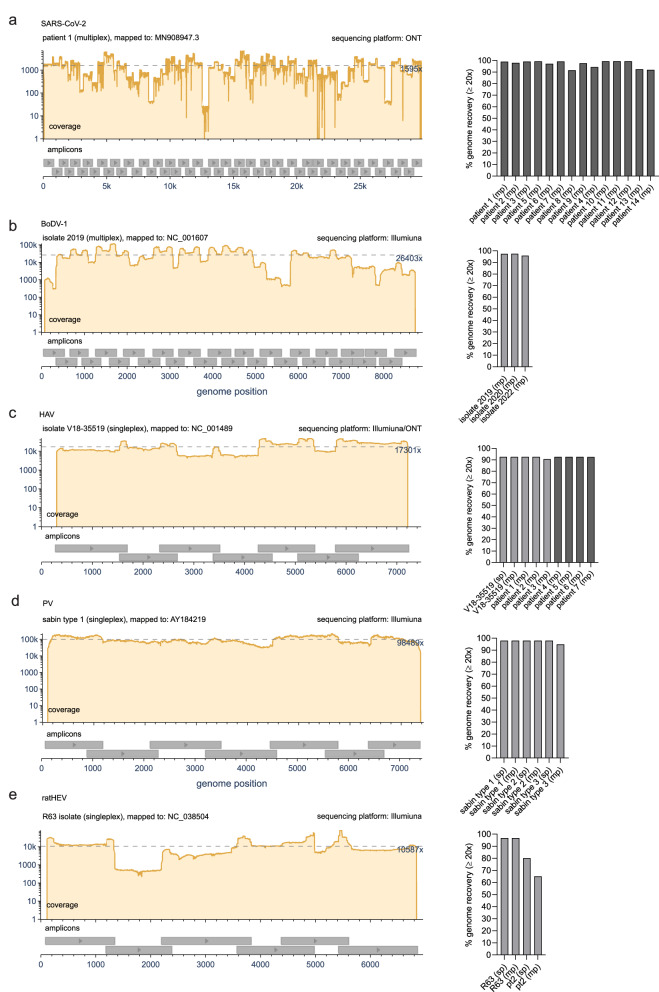
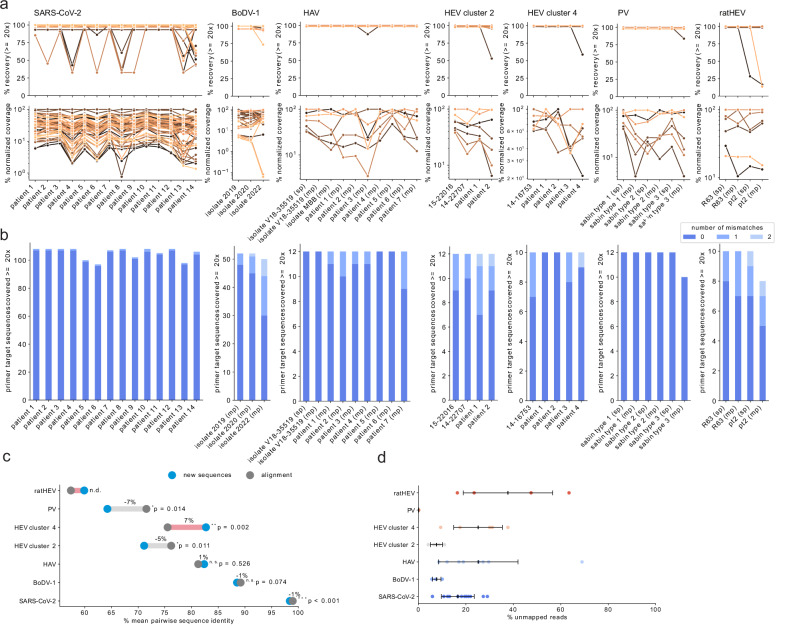
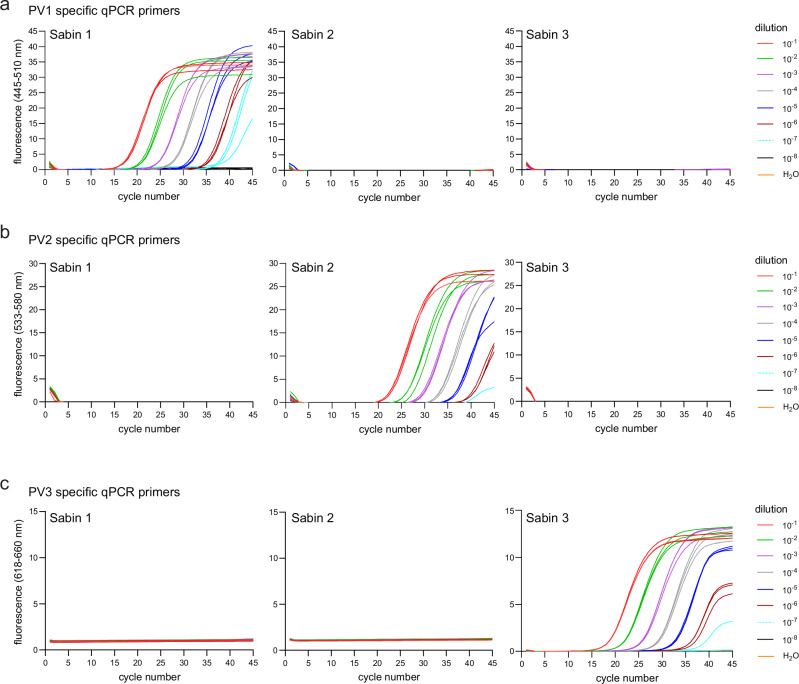
Time- and cost-saving surveillance of viral pathogens is achieved by tiled sequencing in which a viral genome is amplified in overlapping PCR amplicons and qPCR. However, designing pan-specific primers for viral pathogens with high genomic variability represents a significant challenge. Here, we present a bioinformatics command-line tool, called varVAMP (variable virus amplicons), which addresses this issue. It relies on multiple sequence alignments of highly variable virus sequences and enables degenerate primer design for qPCR or tiled amplicon whole genome sequencing. We demonstrate the utility of varVAMP by designing and evaluating novel pan-specific primer schemes suitable for sequencing the genomes of SARS-CoV-2, Hepatitis E virus, rat Hepatitis E virus, Hepatitis A virus, Borna-disease-virus-1, and Poliovirus using clinical samples. Importantly, we also designed primers on the same input data using the software packages PrimalScheme and Olivar and showed that varVAMP minimizes primer mismatches most efficiently. Finally, we established highly sensitive and specific Poliovirus qPCR assays that could potentially simplify current Poliovirus surveillance. varVAMP is open-source and available through PyPI, UseGalaxy, Bioconda, and https://github.com/jonas-fuchs/varVAMP .
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
