

Tenascin-C drives cardiovascular dysfunction in a mouse model of diabetic cardiomyopathy
- PMID: 40450269
- PMCID: PMC12125828
- DOI: 10.1186/s12933-025-02780-y
Tenascin-C drives cardiovascular dysfunction in a mouse model of diabetic cardiomyopathy
Abstract
Background: Diabetic cardiomyopathy (DCM) is a complex condition linked to diabetes, characterized by cardiac and vascular dysfunction, frequently concomitant with heart failure with preserved ejection fraction. The extracellular matrix glycoprotein Tenascin-C (TNC) has been found to be upregulated under diabetic conditions. However, the potential contributory role of TNC in the progression of DCM remains largely unclear. This study was designed to elucidate the role of TNC in the pathogenesis of DCM.
Methods: Diabetes was induced in adult male wild-type (WT) and TNC knockout (TNC-KO) mice, through the administration of streptozotocin (50 mg/kg) for five consecutive days. At 18 weeks cardiac and aortic vascular function was evaluated using echocardiography and wire myography. Myocardium and plasma samples were collected for biochemical, histological, and molecular analyses. Cardiomyocytes and cardiac fibroblasts were used to investigate the impact of diabetes on TNC expression, inflammation, myocardial stiffness and function. Additionally, transcriptomic analysis of cardiac tissue by RNA-sequencing was conducted. Plasma TNC levels were assessed by enzyme-linked immunosorbent assay in cohorts of heart failure patients and type 2 diabetes mellitus.
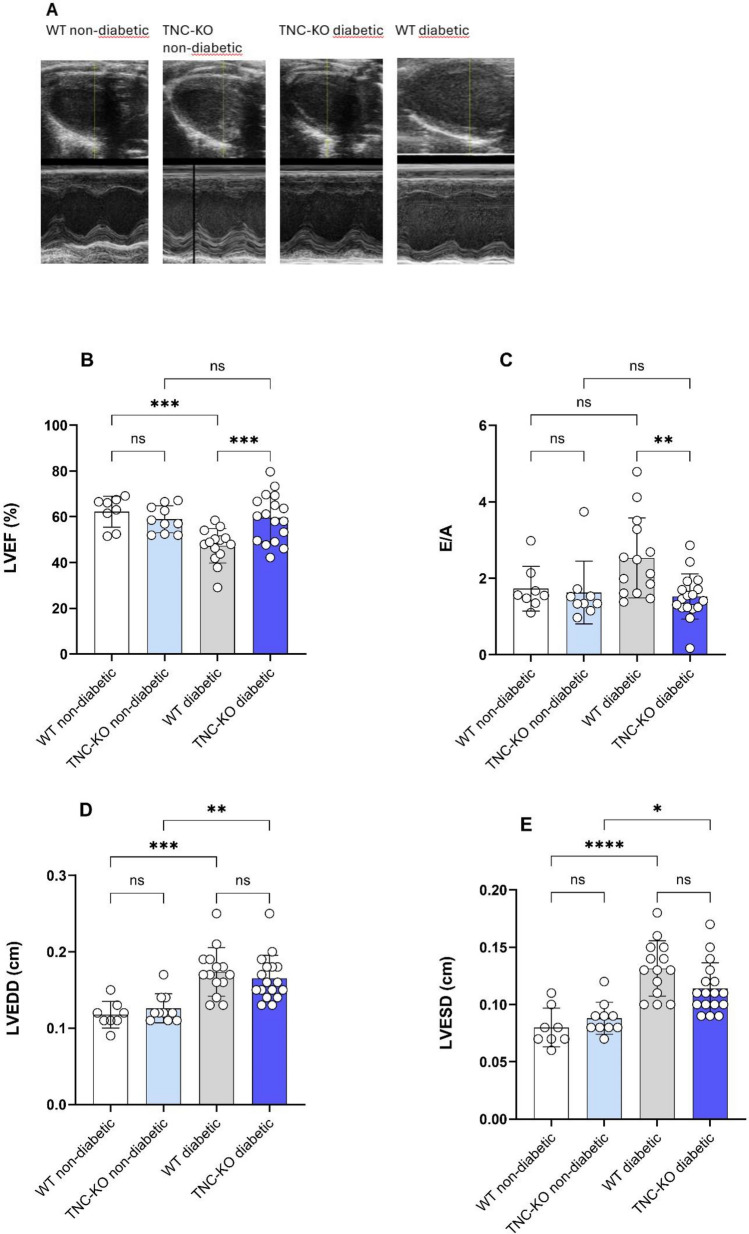
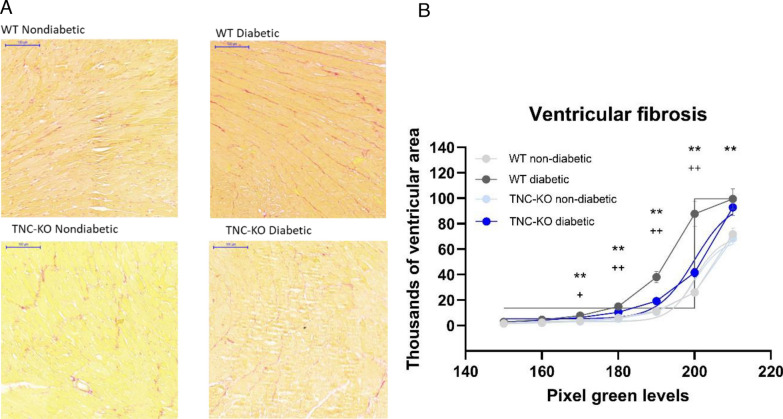
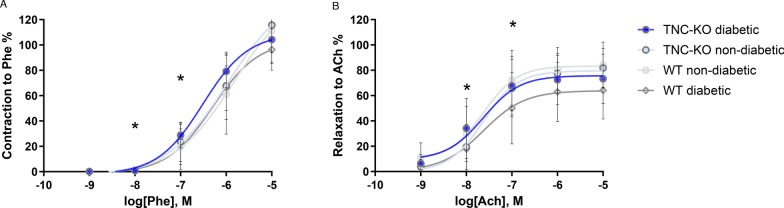
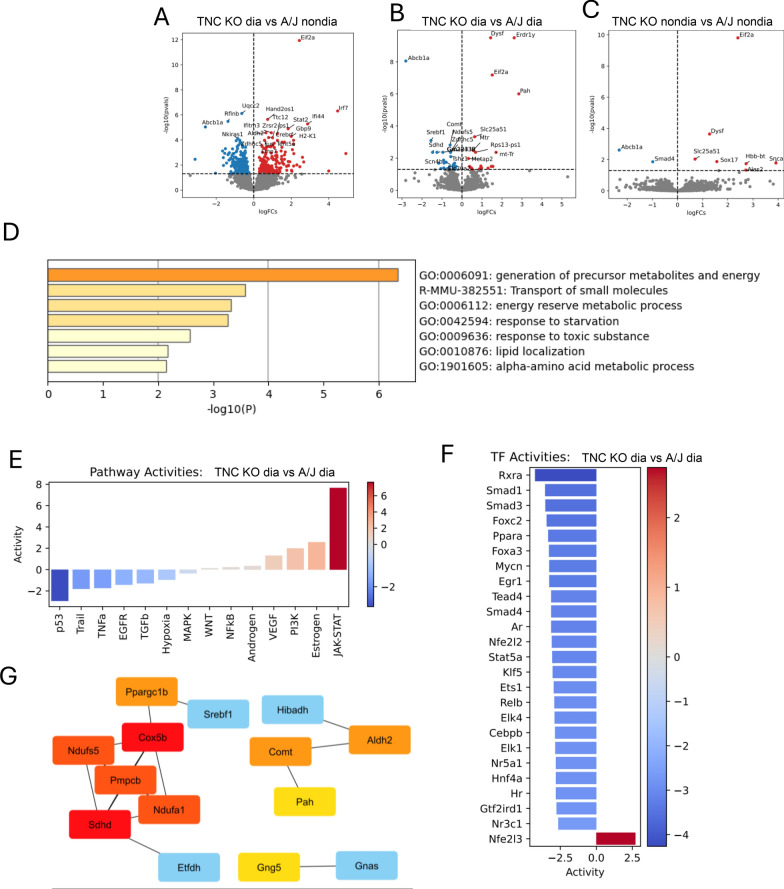
Results: TNC-KO diabetic mice showed preserved left ventricular systolic and diastolic function, significantly reduced cardiac fibrosis and mitigated endothelial dysfunction compared to WT diabetic animals. Compared with cardiomyocytes of diabetic WT animals, cardiomyocytes of TNC-KO mice developed less stiffness (Fpassive). Additionally, exposing mouse cardiomyocytes and human cardiac fibroblasts to high glucose stress (30 mM) led to a significant increase in TNC expression. Conversely, recombinant human TNC promoted pro-inflammatory and oxidative stress markers in cardiomyocytes. The role of TNC in fibrosis and DCM was found to involve pathways related to p53 signaling and Serpin1k, Ccn1, Cpt1a, and Slc27a1, as identified by RNA sequencing analysis. Additionally, plasma TNC levels were significantly elevated in patients with heart failure, irrespective of diabetes status, compared to healthy individuals.
Conclusions: Our findings indicate that in diabetes, TNC contributes to cardiac contractile dysfunction, myocardial fibrosis, oxidative stress, inflammation, and metabolic disturbances in diabetic mouse heart. These results implicate the potential of TNC inhibition as a novel therapeutic approach for treating DCM.
Keywords: Diabetic cardiomyopathy; Fibrosis; Heart failure pathophysiology; Inflammation; Tenascin-C.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: The experimental protocol was approved by the regional Ethics Committee for Laboratory Animal Experiments at the Medical University of Vienna and the Austrian Ministry of Science Research and Economy (BMWFW- 66.009/0014-V/3b/2018). All procedures conform to the guidelines from ARRIVE and Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. The MetAnEnd study was conducted according to the International Conference on Harmonization and Good Clinical Practice guidelines and the Declaration of Helsinki. It was approved by the ethical review board at Karolinksa Institutet (Stockholm, Sweden). Written informed consent was obtained from all participants prior to study participation. Consent for publication: All authors have read and agreed to the publication of this manuscript. Competing interests: The authors declare no competing interests.
Figures






References
-
- Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, Colagiuri S, Guariguata L, Motala AA, Ogurtsova K. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the international diabetes federation diabetes atlas. Diabetes Res Clin Pract. 2019;157:107843. - PubMed
-
- WHO: Cause-specific mortality, 2000–2019. In.; 2020.
-
- Dal Canto E, Ceriello A, Rydén L, Ferrini M, Hansen TB, Schnell O, Standl E, Beulens JW. Diabetes as a cardiovascular risk factor: an overview of global trends of macro and micro vascular complications. Eur J Prev Cardiol. 2019;26(2):25–32. - PubMed
-
- Kannel WB, McGee DL. Diabetes and cardiovascular disease: the Framingham study. JAMA. 1979;241(19):2035–8. - PubMed
-
- Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30(6):595–602. - PubMed
MeSH terms
Substances
Grants and funding
- EFOP-3.6.3-VEKOP-16-2017-00009/EFOP-3.6.3-VEKOP-16-2017-00009
- 523-2014-2336/Karolinska Institutet, the Swedish Research Council
- 20190310, 20220540/Swedish Heart Lung Foundation
- FoUI-974688/Stockholm County Council
- RRF-2.3.1-21-2022-00003/support provided by the European Union
- RRF-2.3.1-21-2022-00003/support provided by the European Union
- Lendület 2024/HUN-REN Hungarian Research Network and from the Semmelweis Lendület 2024 grant
- Lendület 2024/HUN-REN Hungarian Research Network and from the Semmelweis Lendület 2024 grant
- No. 739593/EU's Horizon 2020 research and innovation program under grant agreement
- HA 7512/2-4 and HA 7512/2-1/DFG (Deutsche Forschungsgemeinschaft)
- No. IF-023-22 and No. IF-034-22/InnovationsFoRUM program of the Medical Faculty, RUB
- Project-Number: 21001/Medical-Scientific Fund of the Mayor of the City of Vienna (Project-Number: 21001) ); Austrian-Hungarian Action Foundation project no. 98öu4 and 104öu5 to A.K and Ludwig Boltzmann Institute for Cardiovascular Research (CARREM project line, 2020-2024)
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
