

Sexual epigenetics: genome-wide analysis revealed differential DNA methylation in the vector tick Haemaphysalis longicornis
- PMID: 40452039
- PMCID: PMC12128260
- DOI: 10.1186/s13071-025-06810-2
Sexual epigenetics: genome-wide analysis revealed differential DNA methylation in the vector tick Haemaphysalis longicornis
Abstract
Background: Haemaphysalis longicornis is an important vector that transmits a variety of pathogens to humans and animals. This tick species is unique for having two separate reproductive populations: bisexual and parthenogenetic populations. In bisexual populations, morphological differences exist between the males and females, with the females often larger than the males. DNA methylation, as a key epigenetic modification, plays a crucial role in biological processes such as the maintenance of normal cellular function, the regulation of gene expression, and embryonic development. However, the epigenetic mechanism underlying sex differentiation in the bisexual population of H. longicornis has been overlooked.
Methods: In the present study, the global DNA methylation profiles of the female and male H. longicornis ticks from the bisexual population were explored using whole-genome bisulfite sequencing. Differentially methylated regions (DMRs) were identified, followed by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of DMR-related genes.
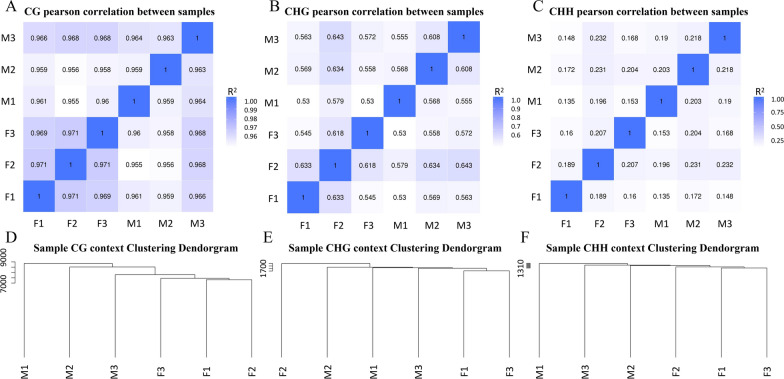
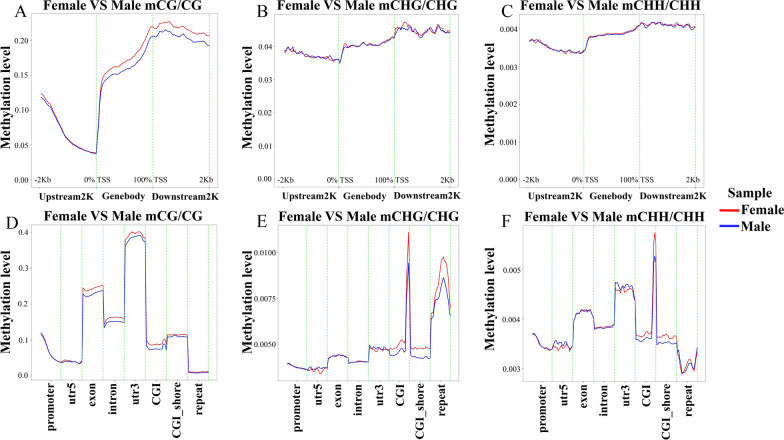
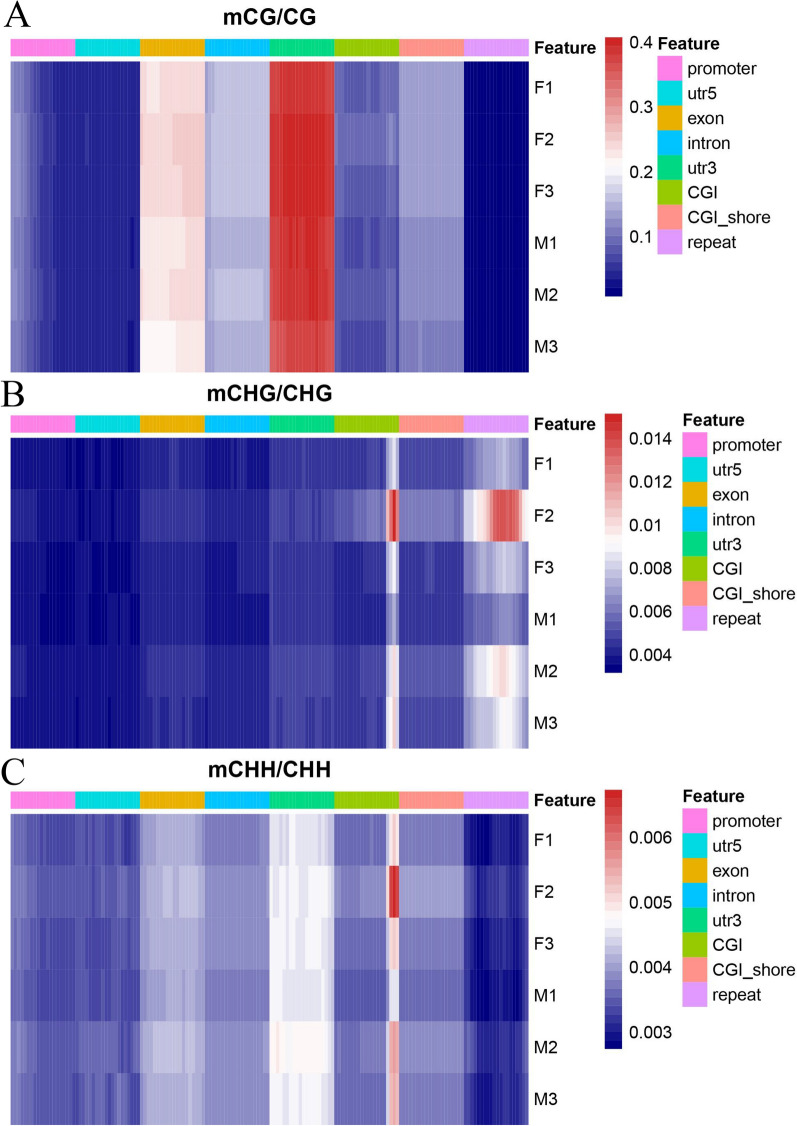
Results: The results revealed that DNA methylation levels in H. longicornis varied by sex and sequence context (CG, CHG, and CHH). The 3' untranslated region (UTR) had the highest methylation in the CG context, followed by exons, introns, and CGI_shore regions. Female ticks generally exhibited higher methylation levels than males, particularly in gene body regions. A total of 10,460 DMRs were identified, with 5282 hypermethylated and 5178 hypomethylated. Further, GO and KEGG pathway analyses showed that differentially methylated genes (DMGs) were highly enriched in binding and metabolic pathways.
Conclusions: These results broaden our understanding of DNA methylation changes associated with the female and male of H. longicornis and provide an important theoretical basis for subsequent studies of epigenetic mechanisms of sex differences in ticks.
Keywords: Haemaphysalis longicornis; DNA methylation; Epigenetic regulation; Sexual dimorphism; Whole-genome bisulfite sequencing.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: All experiments involving rabbits were approved by the Animal Ethics Committee of Hebei Normal University (Protocol Number: IACUC-209230). Competing interests: The authors declare no competing interests. Consent for publication: Not applicable.
Figures



References
-
- Yu X, Marshall H, Liu Y, Xiong Y, Zeng X, Yu H, et al. Sex-specific transcription and DNA methylation landscapes of the Asian citrus psyllid, a vector of huanglongbing pathogens. Evolution. 2023;77:1203–15. 10.1093/evolut/qpad036. - PubMed
-
- Mank JE. Sex chromosomes and the evolution of sexual dimorphism: lessons from the genome. Am Nat. 2009;173:141–50. 10.1086/595754. - PubMed
-
- Bain SA, Marshall H, de la Filia AG, Laetsch DR, Husnik F, Ross L. Sex-specific expression and DNA methylation in a species with extreme sexual dimorphism and paternal genome elimination. Mol Ecol. 2021;30:5687–703. 10.1111/mec.15842. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
