

Polybromo 1/vimentin axis dictates tumor grade, epithelial-mesenchymal transition, and metastasis in pancreatic cancer
- PMID: 40454484
- PMCID: PMC12126225
- DOI: 10.1172/JCI177533
Polybromo 1/vimentin axis dictates tumor grade, epithelial-mesenchymal transition, and metastasis in pancreatic cancer
Abstract
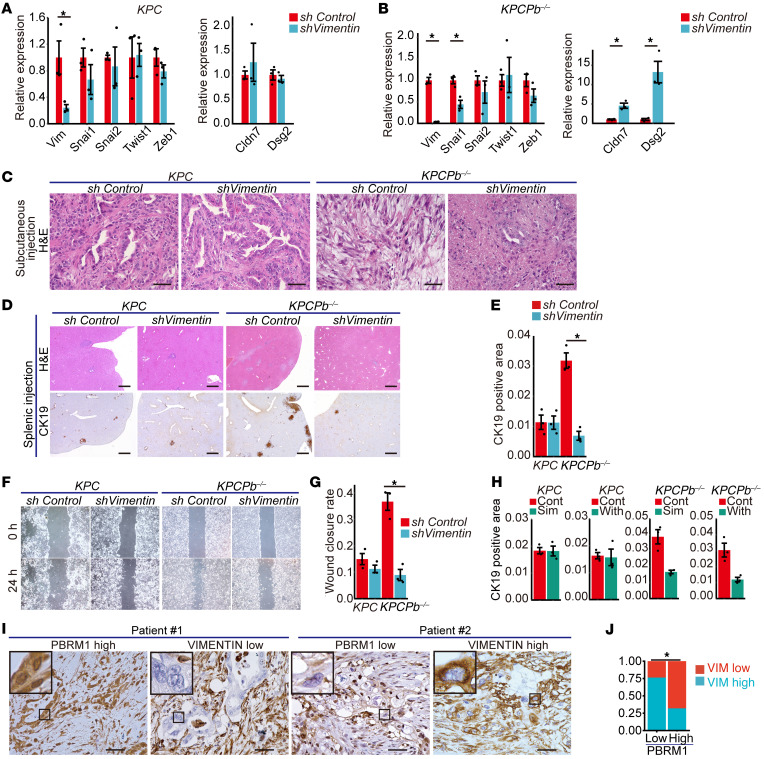
Mutations in Polybromo 1 (PBRM1), a subunit of the switch/sucrose nonfermentable (SWI/SNF) chromatin remodeling complex, are frequently observed in several cancers, including pancreatic ductal adenocarcinoma (PDAC). In this study, we demonstrated that pancreas-specific loss of Pbrm1 in mice harboring Kras mutations and Trp53 deletions accelerated the development of poorly differentiated PDAC, epithelial-mesenchymal transition (EMT), and metastasis, resulting in worsened prognosis. Pbrm1 loss in preexisting PDAC shifted the tumor grade from a well- to a poorly differentiated state and elevated vimentin expression. Pbrm1-null PDAC exhibited downregulation of apical junction genes and upregulation of EMT pathway genes, including the vimentin and squamous molecular subtype signature genes. Mechanistically, PBRM1 bound to the vimentin gene promoter and directly downregulated its expression. Furthermore, suppression of vimentin in Pbrm1-null PDAC cells reversed the dedifferentiation phenotype and reduced EMT and metastasis. Consistently, reduced PBRM1 expression correlated with high vimentin expression, poorly differentiated histology, a high recurrence rate, and reduced overall survival in human PDACs. Additionally, PDAC with PBRM1 deletion was associated with the aggressive squamous molecular subtype. Our data established PBRM1 as a tumor suppressor that controls tumor grade and metastasis of PDAC by regulating vimentin expression.
Keywords: Cancer; Epigenetics; Gastroenterology; Mouse models; Oncology.
Conflict of interest statement
Figures






Comment in
- Switching on the evolutionary potential of pancreatic cancer: the tumor suppressor functions of PBRM1 doi: 10.1172/JCI193205
References
-
- Bednar F, Di Magliano MP. Chemotherapy and tumor evolution shape pancreatic cancer recurrence after resection. Cancer Discov. 2020;10(6):762–764. doi: 10.1158/2159-8290.CD-20-0359. - DOI - PubMed
-
- Herman JM, et al. Analysis of fluorouracil-based adjuvant chemotherapy and radiation after pancreaticoduodenectomy for ductal adenocarcinoma of the pancreas: results of a large, prospectively collected database at the Johns Hopkins Hospital. J Clin Oncol. 2008;26(21):3503–3510. doi: 10.1200/JCO.2007.15.8469. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
