

Myocardial mitochondrial antiviral signaling protein promotes heart Ischemia-reperfusion injury via RIG-I signaling in mice
- PMID: 40456736
- PMCID: PMC12130335
- DOI: 10.1038/s41467-025-60123-7
Myocardial mitochondrial antiviral signaling protein promotes heart Ischemia-reperfusion injury via RIG-I signaling in mice
Abstract
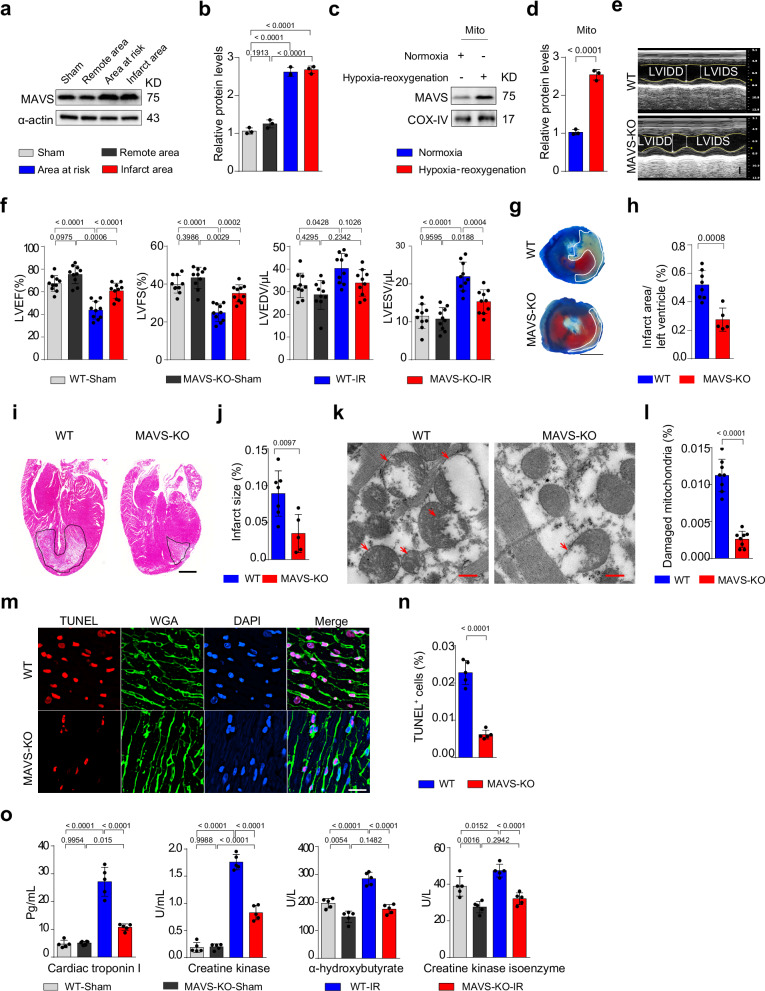
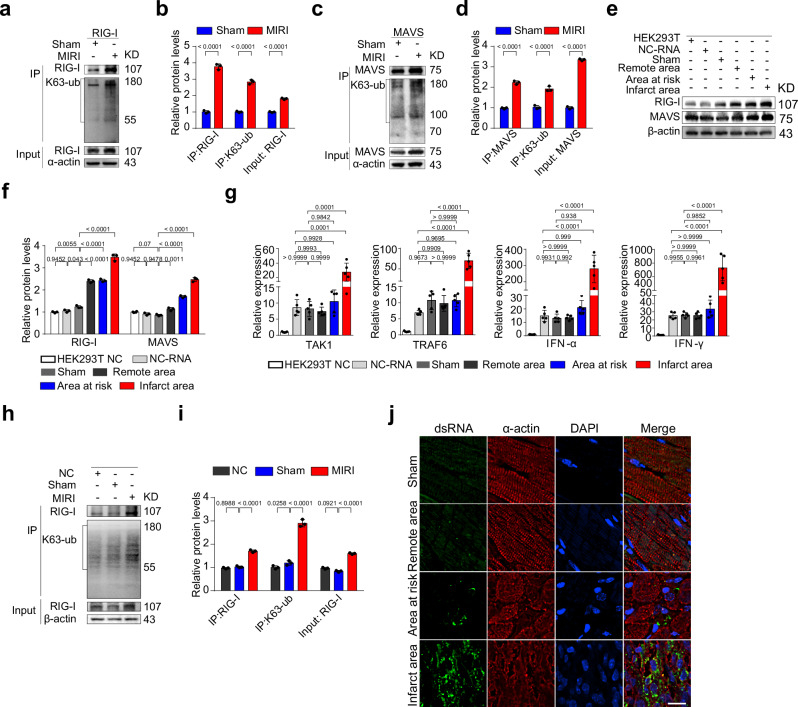
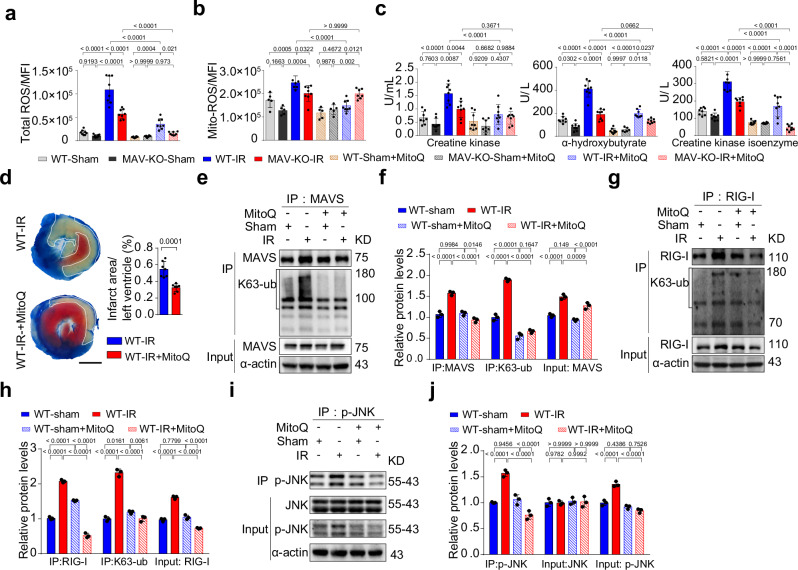
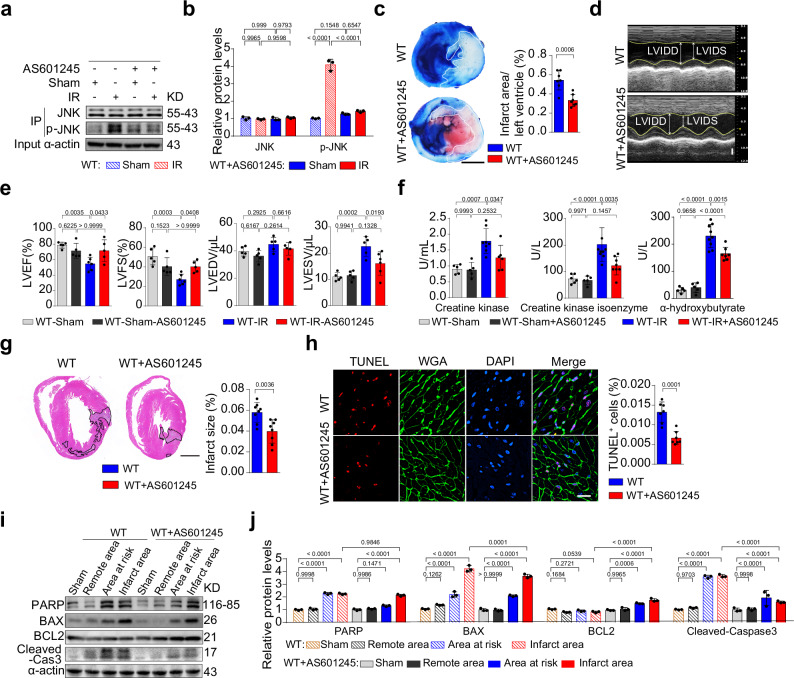
Myocardial ischemia-reperfusion injury (MIRI) is a life-threatening complication of myocardial infarcts, with inner mitochondrial membrane protein dysfunction involved in MIRI-induced heart injury. The role of outer mitochondrial membrane protein mitochondrial antiviral signaling protein (MAVS) is unknown. Here, we show that MAVS expression increases in infarcted myocardium of male wild-type mice. Global MAVS-knock-out or myocardial-specific MAVS knockdown protects male mice from acute and chronic MIRI. MIRI induces double-stranded RNA in affected myocardium, activating intracellular retinoic acid-inducible gene I (RIG-I) signaling, which leads to MAVS aggregation and subsequent non-canonical downstream signaling. MAVS aggregates recruit tumor necrosis factor-associated factor family 6 (TRAF6) and transforming growth factor-β-activated kinase 1 (TAK1), the activating mitogen-activated protein kinase (MAPK) pathway and apoptosis. MAVS-knock-out reduces c-jun-NH2 terminal kinase (JNK) phosphorylation and apoptosis. JNK inhibition protects against MIRI in wild-type male mice, whereas JNK agonist impairs protection in MAVS-knock-out male mice. MIRI activates RIG-I/MAVS pathway and subsequently triggers the TAK1/TRAF6 complex, leading to the activation of the MAPK/JNK signaling cascade. This sequential activation cascade may serve as a potential therapeutic target for MIRI.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures





References
-
- Ibáñez, B., Heusch, G., Ovize, M. & Van de Werf, F. Evolving therapies for myocardial ischemia/reperfusion injury. J. Am. Coll. Cardiol.65, 1454–1471 (2015). - PubMed
-
- Heusch, G. Cardioprotection research must leave its comfort zone. Eur. Heart J.39, 3393–3395 (2018). - PubMed
-
- Hausenloy, D. J. & Yellon, D. M. Ischaemic conditioning and reperfusion injury. Nat. Rev. Cardiol.13, 193–209 (2016). - PubMed
-
- Jernberg, T. et al. Association between adoption of evidence-based treatment and survival for patients with ST-elevation myocardial infarction. Jama305, 1677–1684 (2011). - PubMed
MeSH terms
Substances
Grants and funding
- HA 1083/15-5/Deutsche Forschungsgemeinschaft (German Research Foundation)
- Plaquefight 01KL1808/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
- 82274317/National Natural Science Foundation of China (National Science Foundation of China)
- 82474287/National Natural Science Foundation of China (National Science Foundation of China)
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
