

Phylogenetic inference reveals clonal heterogeneity in circulating tumor cell clusters
- PMID: 40457073
- PMCID: PMC12165839
- DOI: 10.1038/s41588-025-02205-2
Phylogenetic inference reveals clonal heterogeneity in circulating tumor cell clusters
Abstract
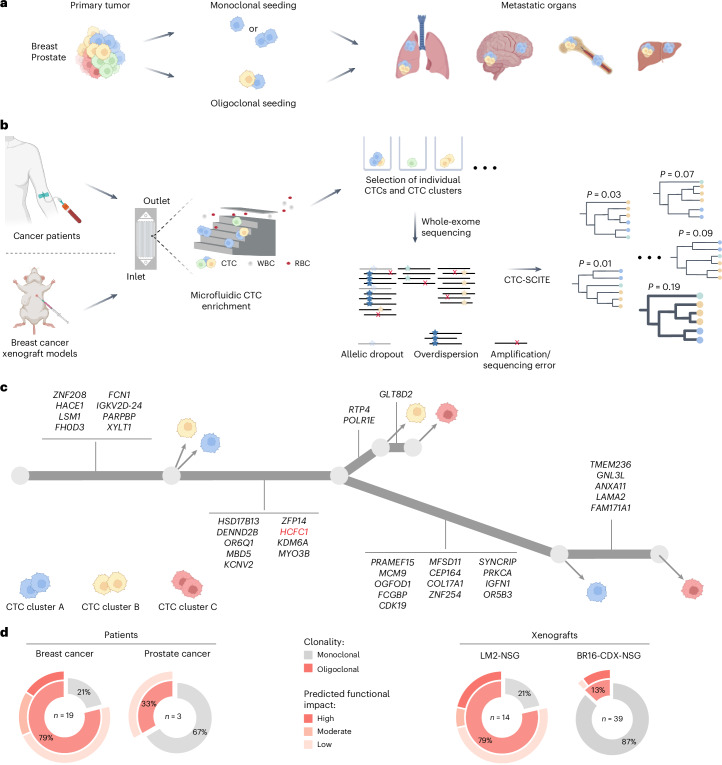
Circulating tumor cell (CTC) clusters are highly efficient metastatic seeds in various cancers. Yet, their genetic heterogeneity and clonal architecture is poorly characterized. Using whole-exome sequencing coupled with phylogenetic inference from CTC clusters of patients with breast and prostate cancer, as well as mouse cancer models alongside barcode-mediated clonal tracking in vivo, we demonstrate oligoclonal composition of individual CTC clusters. These results improve our understanding of metastasis-relevant clonal dynamics.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: N.A. is a cofounder and member of the board of PAGE Therapeutics AG, listed as an inventor in patent applications related to CTCs, a paid consultant for companies with an interest in liquid biopsies and a Novartis shareholder. C.R. is a cofounder, employee and member of the board of PAGE Therapeutics AG. The other authors declare no competing interests.
Figures








References
-
- Llofta, L. A., Klelnerman, J. & Saldel, G. M. The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res.36, 889–894 (1976). - PubMed
MeSH terms
Grants and funding
- 101001652/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
- 212183/WT_/Wellcome Trust/United Kingdom
- KLS-5636-08-2022/Krebsliga Schweiz (Ligue Suisse Contre le Cancer)
- 212183/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
