

Sequential emergence and contraction of epithelial subtypes in the prenatal human choroid plexus revealed by a stem cell model
- PMID: 40461502
- PMCID: PMC12134268
- DOI: 10.1038/s41467-025-60361-9
Sequential emergence and contraction of epithelial subtypes in the prenatal human choroid plexus revealed by a stem cell model
Abstract
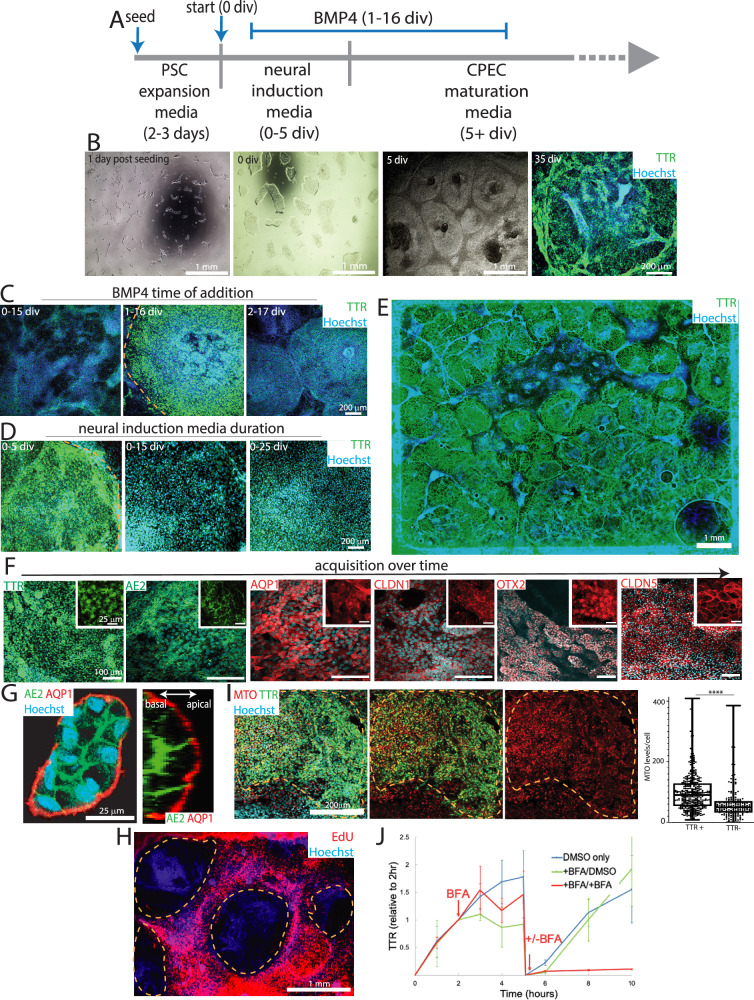
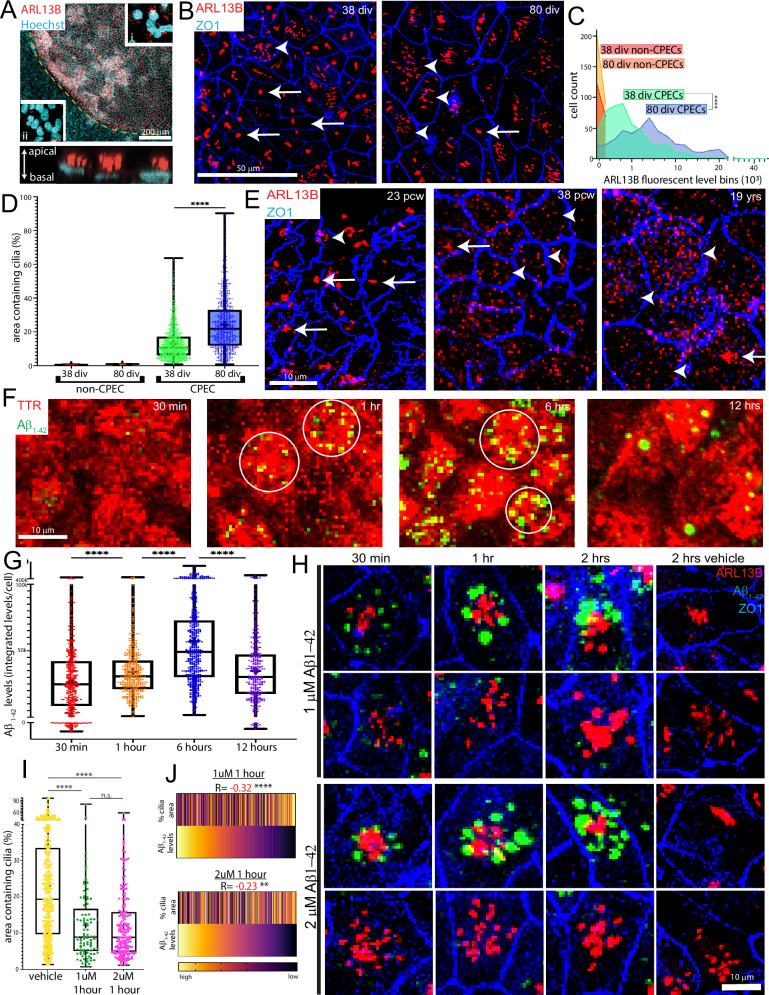
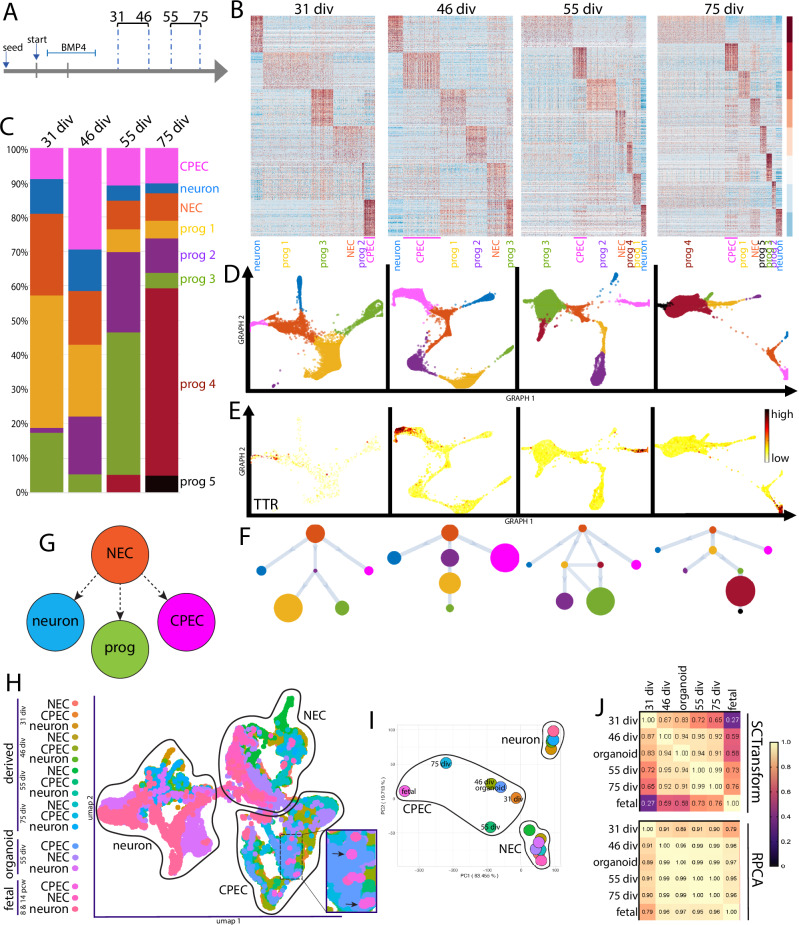
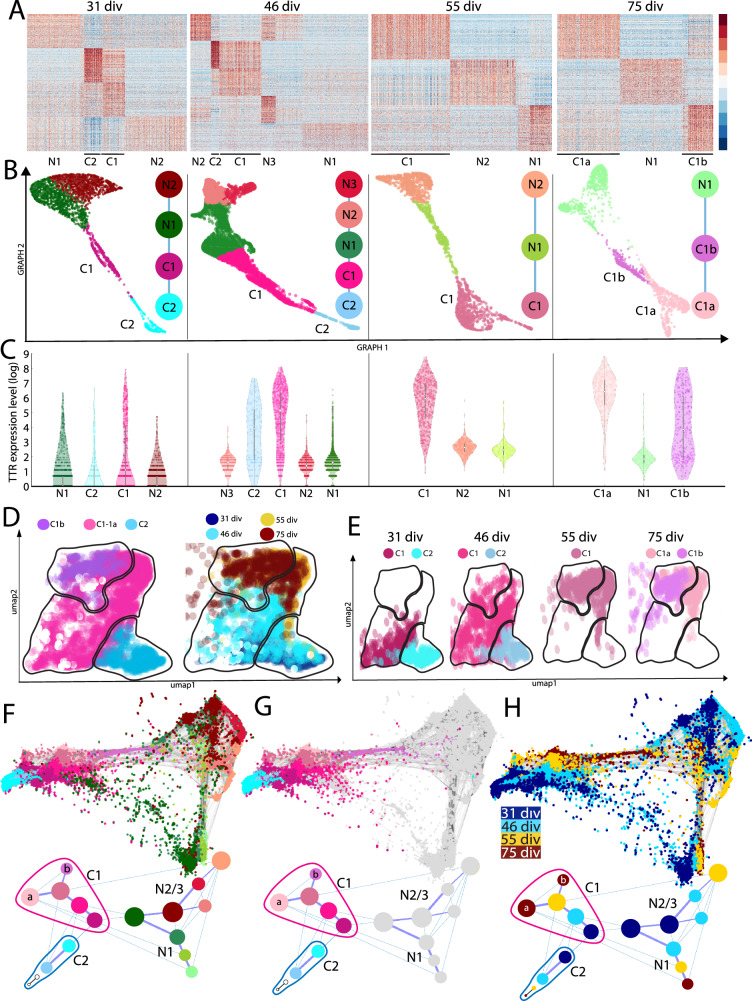
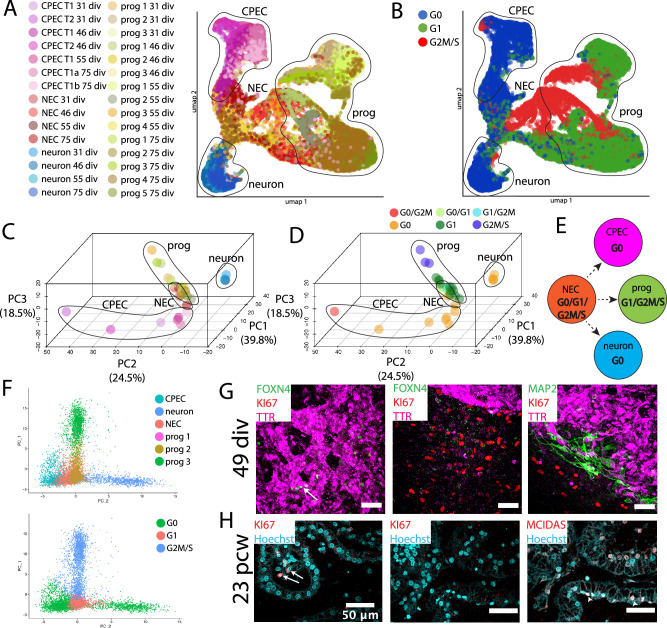
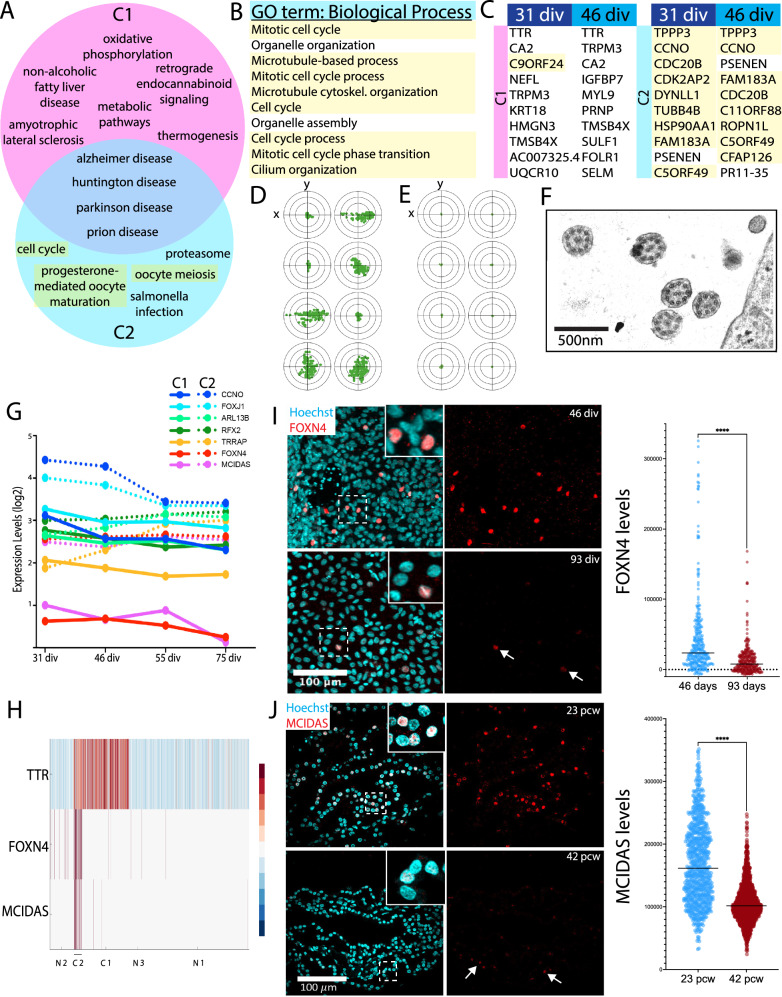
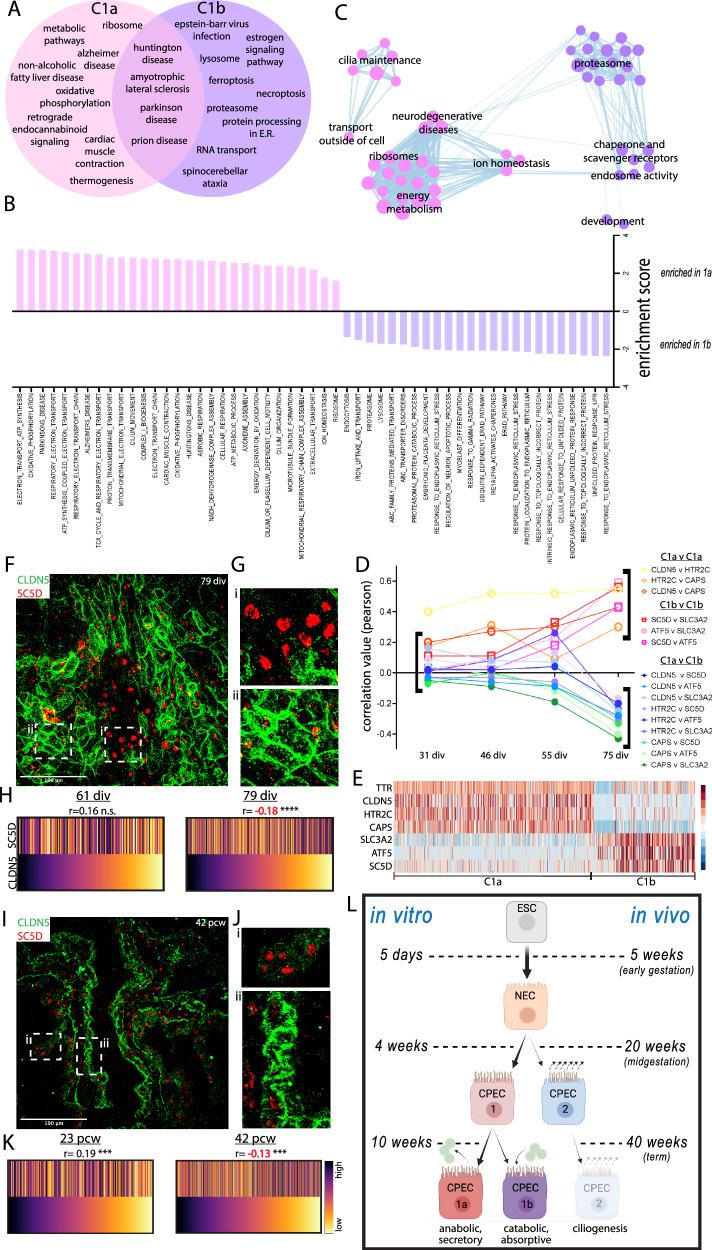
Despite the major roles of choroid plexus epithelial cells (CPECs) in brain homeostasis and repair, their developmental lineage and diversity remain undefined. In simplified differentiations from human pluripotent stem cells, derived CPECs (dCPECs) display canonical properties and dynamic motile multiciliated phenotypes that interact with Aβ uptake. Single dCPEC transcriptomes over time correlate well with human organoid and fetal CPECs, while pseudotemporal and cell cycle analyses highlight the direct CPEC origin from neuroepithelial cells. In addition, time series analyses define metabolic (type 1) and ciliogenic dCPECs (type 2) at early timepoints, followed by type 1 diversification into anabolic-secretory (type 1a) and catabolic-absorptive subtypes (type 1b) as type 2 cells contract. These temporal patterns are then confirmed in independent derivations and mapped to prenatal stages using human tissues. In addition to defining the prenatal lineage of human CPECs, these findings suggest dynamic models of ChP support for the developing human brain.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Update of
-
Sequential emergence and contraction of epithelial subtypes in the prenatal human choroid plexus revealed by a stem cell model.bioRxiv [Preprint]. 2024 Jun 17:2024.06.12.598747. doi: 10.1101/2024.06.12.598747. bioRxiv. 2024. Update in: Nat Commun. 2025 Jun 3;16(1):5149. doi: 10.1038/s41467-025-60361-9. PMID: 38948782 Free PMC article. Updated. Preprint.
References
-
- Faraci, F. M., Mayhan, W. G. & Heistad, D. D. Effect of serotonin on blood flow to the choroid plexus. Brain Res.478, 121–126 (1989). - PubMed
-
- Faraci, F. M., Baumbach, G. L. & Heistad, D. D. Cerebral circulation: humoral regulation and effects of chronic hypertension. J. Am. Soc. Nephrol.1, 53–57 (1990). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
