

Pooled analysis of 3,741 stool metagenomes from 18 cohorts for cross-stage and strain-level reproducible microbial biomarkers of colorectal cancer
- PMID: 40461820
- PMCID: PMC12283368
- DOI: 10.1038/s41591-025-03693-9
Pooled analysis of 3,741 stool metagenomes from 18 cohorts for cross-stage and strain-level reproducible microbial biomarkers of colorectal cancer
Abstract
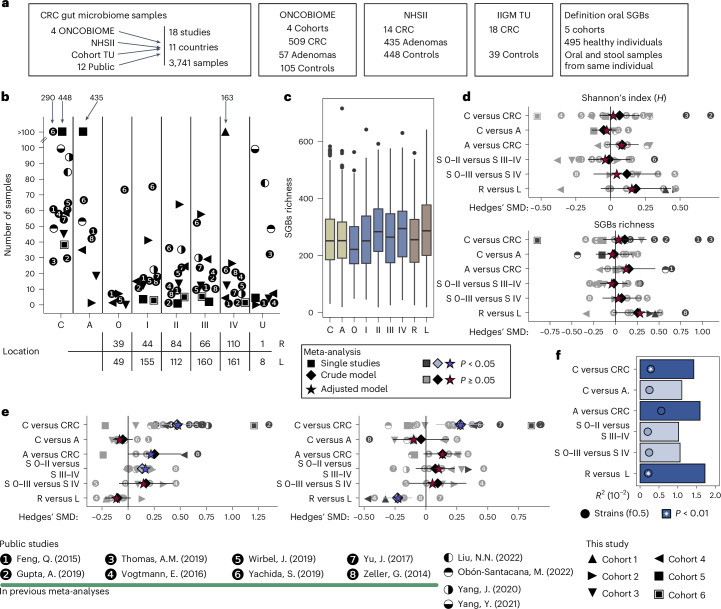
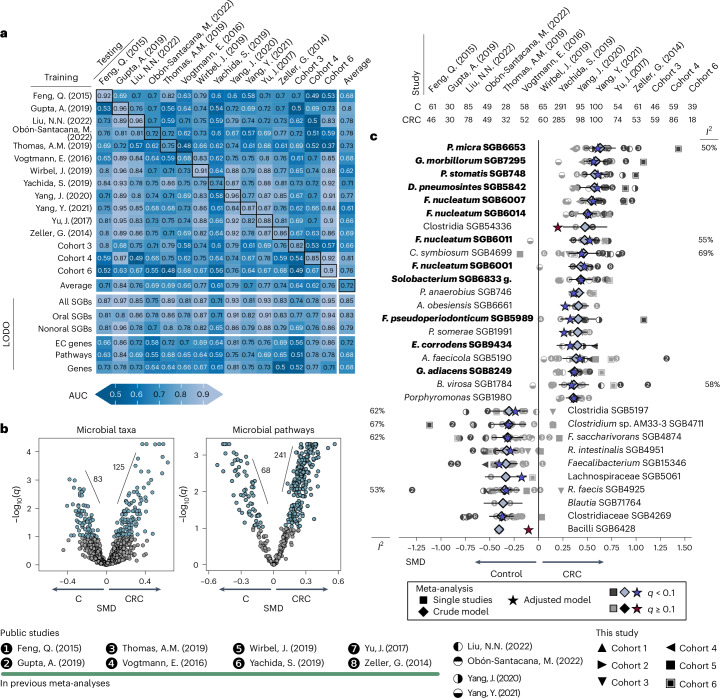
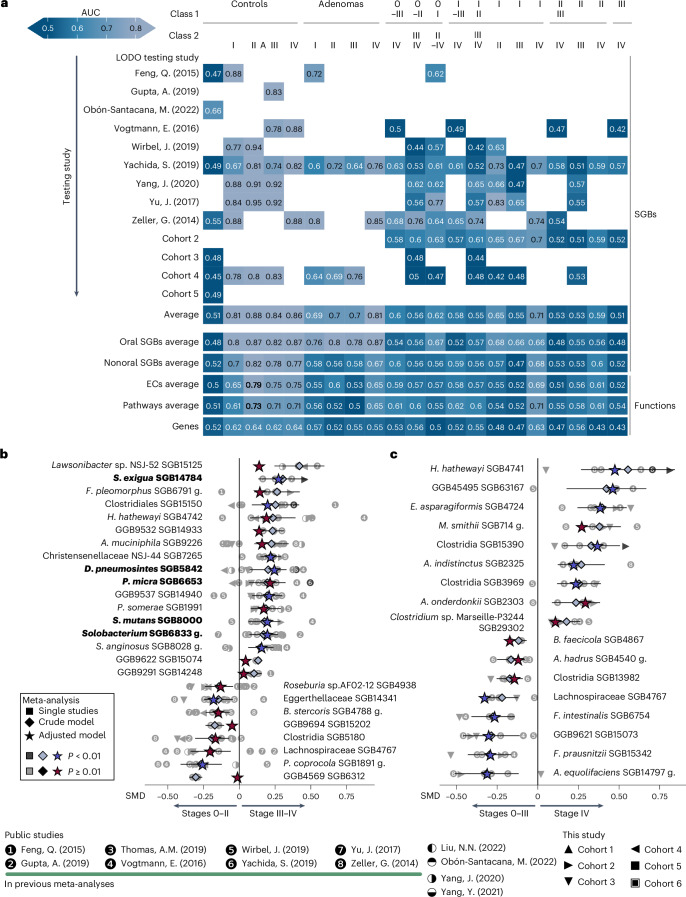
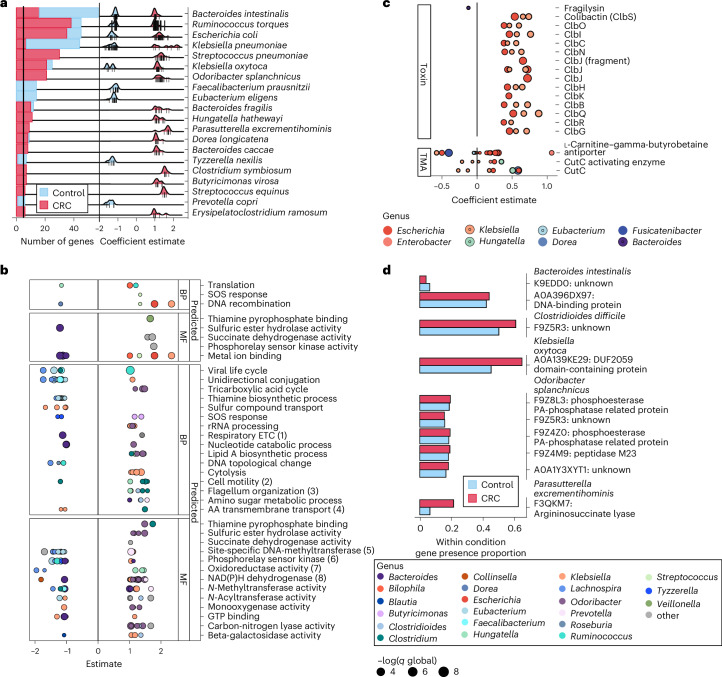
Associations between the gut microbiome and colorectal cancer (CRC) have been uncovered, but larger and more diverse studies are needed to assess their potential clinical use. We expanded upon 12 metagenomic datasets of patients with CRC (n = 930), adenomas (n = 210) and healthy control individuals (n = 976; total n = 2,116) with 6 new cohorts (n = 1,625) providing granular information on cancer stage and the anatomic location of tumors. We improved CRC prediction accuracy based solely on gut metagenomics (average area under the curve = 0.85) and highlighted the contribution of 19 newly profiled species and distinct Fusobacterium nucleatum clades. Specific gut species distinguish left-sided versus right-sided CRC (area under the curve = 0.66) with an enrichment of oral-typical microbes. We identified strain-specific CRC signatures with the commensal Ruminococcus bicirculans and Faecalibacterium prausnitzii showing subclades associated with late-stage CRC. Our analysis confirms that the microbiome can be a clinical target for CRC screening and characterizes it as a biomarker for CRC progression.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: N.S. is a founder and shareholder of PreBiomics Srl and is on the scientific advisory board of ZOE Ltd and received consultancy fees from them. C.H. is a member of the Seres Therapeutics and Empress Therapeutics scientific advisory boards. L.Z. is a founder of Biotech. Cie everImmune involved in the cancer/microbiome space, the president of everImmune scientific advisory board (SAB) and received honoraria from everImmune. L.Z. received research contract fundings from Daiichi Sankyo and Biomérieux. The other authors declare no competing interests.
Figures












References
-
- Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin.71, 209–249 (2021). - PubMed
-
- Siegel, R. L., Wagle, N. S., Cercek, A., Smith, R. A. & Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin.73, 233–254 (2023). - PubMed
-
- Keum, N. & Giovannucci, E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol.16, 713–732 (2019). - PubMed
-
- Miller, K. D. et al. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin.72, 409–436 (2022). - PubMed
MeSH terms
Substances
Grants and funding
- CGCATF-2023/100041/Cancer Research UK (CRUK)
- U01 CA176726/CA/NCI NIH HHS/United States
- OT2 CA297205/CA/NCI NIH HHS/United States
- 825410/EC | Horizon 2020 Framework Programme (EU Framework Programme for Research and Innovation H2020)
- 1OT2CA297205-01/U.S. Department of Health & Human Services | NIH | NCI | Division of Cancer Epidemiology and Genetics, National Cancer Institute (National Cancer Institute Division of Cancer Epidemiology and Genetics)
- R35 CA253185/CA/NCI NIH HHS/United States
- U01 CA230551/CA/NCI NIH HHS/United States
- 101045015/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
- OT2 CA297680/CA/NCI NIH HHS/United States
- U01 CA261961/CA/NCI NIH HHS/United States
- R01 CA263776/CA/NCI NIH HHS/United States
- CGCATF-2023/100036/Cancer Research UK (CRUK)
- OT2CA297680/U.S. Department of Health & Human Services | NIH | NCI | Division of Cancer Epidemiology and Genetics, National Cancer Institute (National Cancer Institute Division of Cancer Epidemiology and Genetics)
LinkOut - more resources
Full Text Sources
Medical
