

Reprogrammed immuno-metabolic environment of cancer: the driving force of ferroptosis resistance
- PMID: 40462094
- PMCID: PMC12131403
- DOI: 10.1186/s12943-025-02337-3
Reprogrammed immuno-metabolic environment of cancer: the driving force of ferroptosis resistance
Abstract
Ferroptosis, the non-apoptotic, iron-dependent form of cell death is an unavoidable outcome and byproduct of cellular metabolism. Reactive oxygen species generation during metabolic activities transcends to Fe2+-induced lipid peroxidation, leading to ferroptosis. Cancer cells being highly metabolic are more prone to ferroptosis. However, their neoplastic nature enables them to bypass ferroptosis and become ferroptosis-resistant. The capability of cancer cells to reprogram its metabolic activities is one of its finest abilities to abort oxidative damage, and hence ferroptosis. Moreover, the reprogrammed metabolism of cancer cells, also associates with the radical trapping antioxidant systems to enhance the scavenging of ferroptosis and thereby tumor progression. Additionally, the TME, which is an inevitable part and regulator of carcinogenesis, presents an intricate cooperation with tumor metabolism to build an immuno-metabolic environment to regulate the sustenance of cell proliferation and survival. This review focuses on the current understanding of ferroptosis in carcinogenesis and its resistance acquired by cancer cells via several modulators including the radical trapping antioxidant systems, the reprogrammed metabolism, the TME, and intertwined role of cancer metabolism and tumor immunity. The reprogrammed metabolism section further comprehends the functional role of lipids, iron and glucose metabolism against ferroptosis defense separately. The affiliation of TME in ferroptosis regulation is further sectioned with reference to different immune cells present within the TME such as tumor-associated macrophages, tumor-infiltrating neutrophils, myeloid-derived suppressor cells, T-cells, natural killer cells, dendritic cells, and B-cells, modifying the TME in both pro and anti-tumorigenic manner. Subsequently, this review also discusses the convergence of immuno-metabolic environment in ferroptosis regulation, and eventually brings up research gaps in this context providing consequential and significant questions to explore for better understanding of the immuno-metabolic environment's role in driving ferroptosis resistance for anti-cancer treatment progress.
Keywords: Ferroptosis; Ferroptosis resistance; Metabolic reprogramming; Reprogrammed immune metabolic environment; Tumor microenvironment.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures
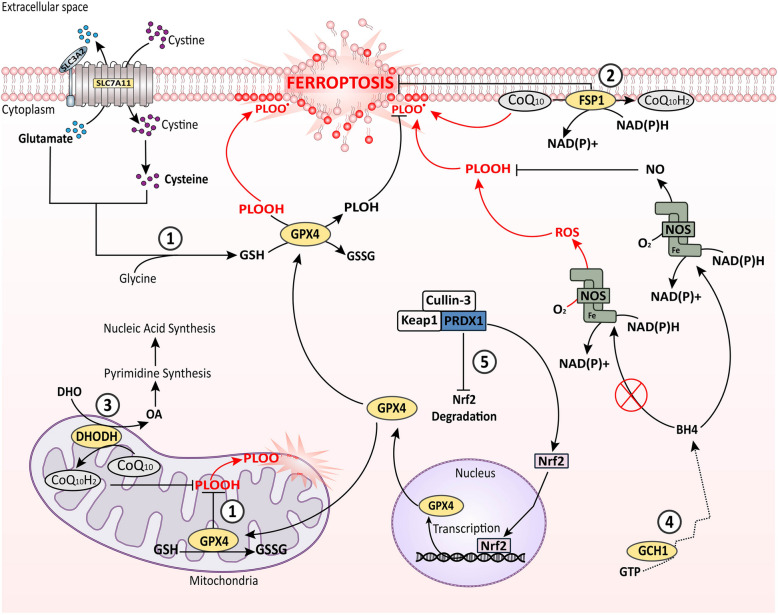
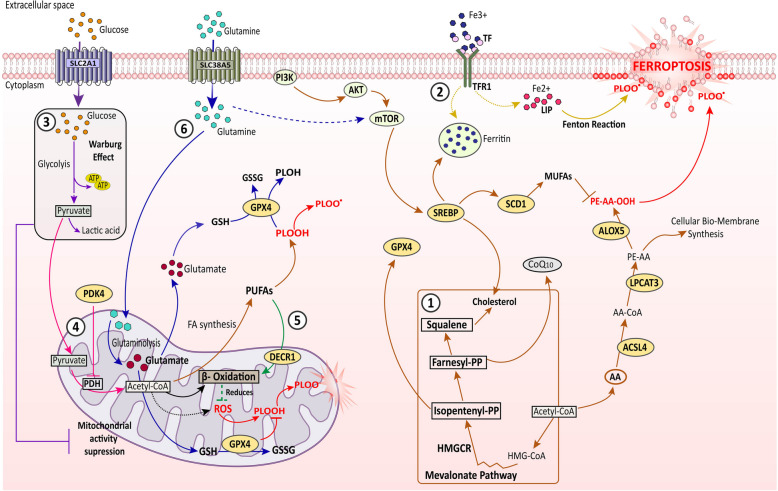
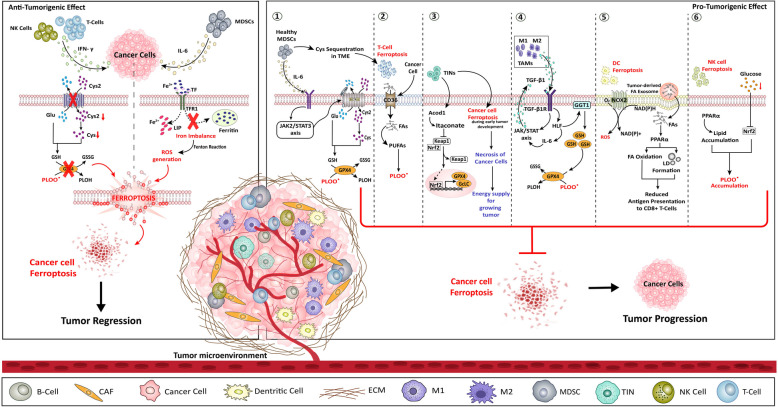
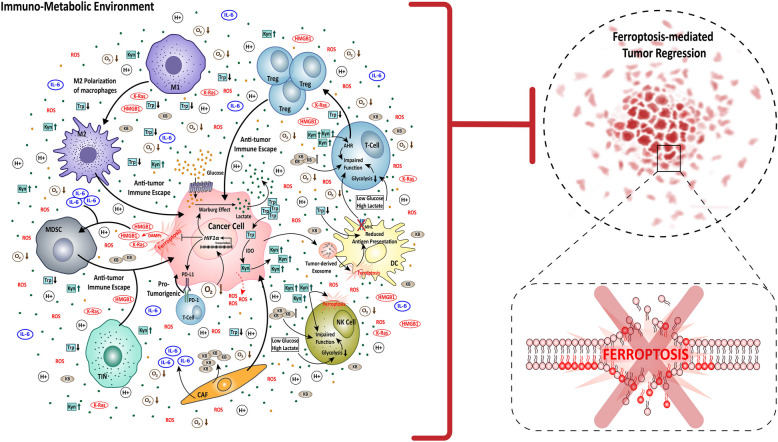
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
