

[PLCE1 mutation-induced end-stage renal disease presenting with massive proteinuria: a family analysis and literature review]
- PMID: 40462432
- PMCID: PMC12169886
- DOI: 10.7499/j.issn.1008-8830.2411029
[PLCE1 mutation-induced end-stage renal disease presenting with massive proteinuria: a family analysis and literature review]
Abstract
Objectives: To summarize the clinical and genetic characteristics of end-stage renal disease caused by PLCE1 gene mutations.
Methods: A retrospective analysis of the clinical and genetic features of three children from a family with PLCE1 gene mutations was conducted, along with a literature review of hereditary kidney disease cases caused by PLCE1 gene mutations.
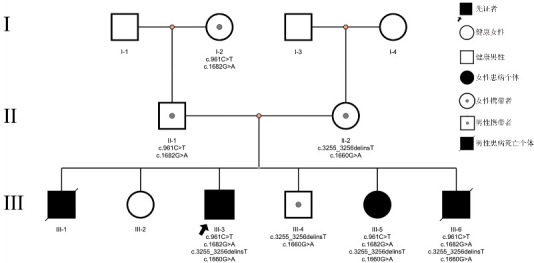
Results: The proband was an 8-year-old male presenting with nephrotic syndrome stage 4 chronic kidney disease. Renal biopsy showed focal segmental glomerulosclerosis. Two years and five months after kidney transplantation, the patient had persistent negative proteinuria and normal renal function. Whole-exome sequencing identified two pathogenic heterozygous variants: c.961C>T and c.3255_3256delinsT, with c.3255_3256delinsT being a novel mutation. Family screening revealed no renal involvement in the parents, but among five siblings, one brother died at age of 4 years from end-stage renal disease. A 7-year-old sister presented with proteinuria and bilateral medullary sponge kidney, with proteinuria resolving after one year of follow-up. A 3-year-old brother died after kidney transplantation due to severe pneumonia. The literature review included 45 patients with hereditary kidney disease caused by PLCE1 gene mutations. The main clinical phenotype was nephrotic syndrome (87%, 39/45), and renal pathology predominantly showed focal segmental glomerulosclerosis (57%, 16/28). No mutation hotspots were identified.
Conclusions: Compound heterozygous mutations in the PLCE1 gene can lead to rapid progression of the disease to end-stage renal disease, with favorable outcomes following kidney transplantation. Family screening is crucial for early diagnosis, and medullary sponge kidney may be a novel phenotype associated with these gene mutations.
目的: 总结PLCE1基因突变致终末期肾病的临床和基因变异特征。方法: 回顾性分析一家系3例PLCE1基因突变患儿的临床和遗传学特征,并对PLCE1基因突变致遗传性肾病病例进行文献复习。结果: 先证者8岁男性,表现为肾病综合征、慢性肾脏病4期,肾活检为局灶性节段性肾小球硬化,肾移植术后2年5个月,尿蛋白持续阴性,肾功能正常。全外显子组测序发现2个致病杂合变异c.961C>T和c.3255_3256delinsT,其中c.3255_3256delinsT为新发突变。家系筛查显示父母无肾脏受累,5位同胞中,1位胞兄于4岁时因终末期肾病死亡;7岁胞妹有蛋白尿和双侧髓质海绵肾,随访1年尿蛋白转阴;3岁胞弟肾移植后因重症肺炎死亡。文献复习共纳入45例PLCE1基因突变致遗传性肾病患者,主要临床表型为肾病综合征(87%,39/45),肾脏病理以局灶性节段性肾小球硬化为主(57%,16/28)。未见热点突变位点。结论: PLCE1基因复合杂合突变可导致疾病迅速进展至终末期肾病,肾移植效果良好,家系筛查对早期诊断至关重要,髓质海绵肾可能为该基因突变的新表型。.
Keywords: Child; End-stage renal disease; Kidney transplantation; Medullary sponge kidney; PLCE1 gene.
Conflict of interest statement
所有作者声明无利益冲突。
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical