

This is a preprint.
Multiomic Evidence for a Unified Model of Alzheimer's Disease Etiology Linking Microglial Flux Capacity and Astrocyte-Neuron Metabolic Breakdown
- PMID: 40462904
- PMCID: PMC12132299
- DOI: 10.1101/2024.07.23.604835
Multiomic Evidence for a Unified Model of Alzheimer's Disease Etiology Linking Microglial Flux Capacity and Astrocyte-Neuron Metabolic Breakdown
Abstract
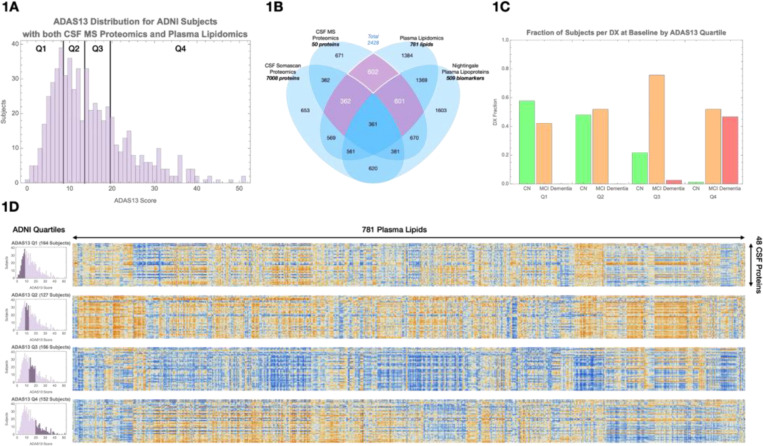
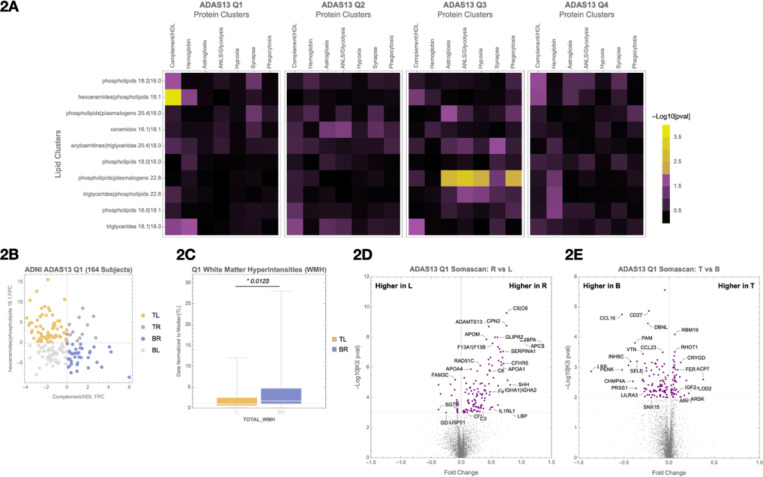
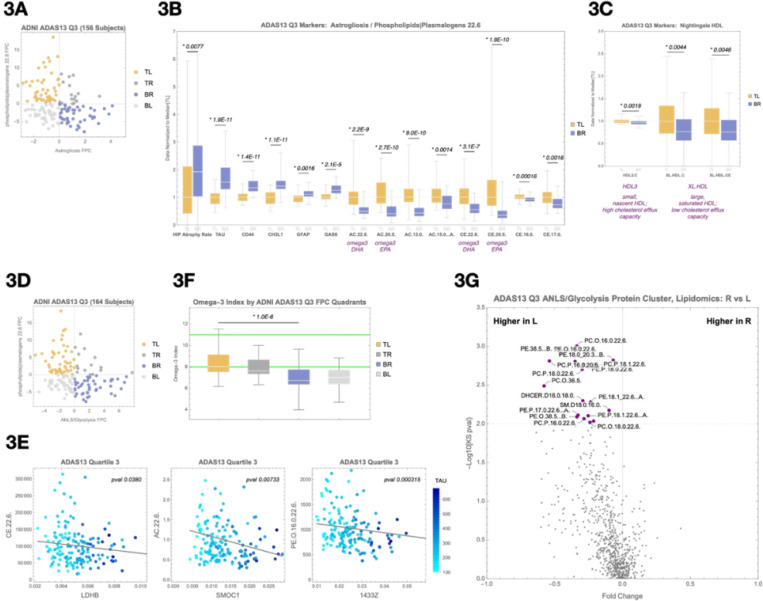
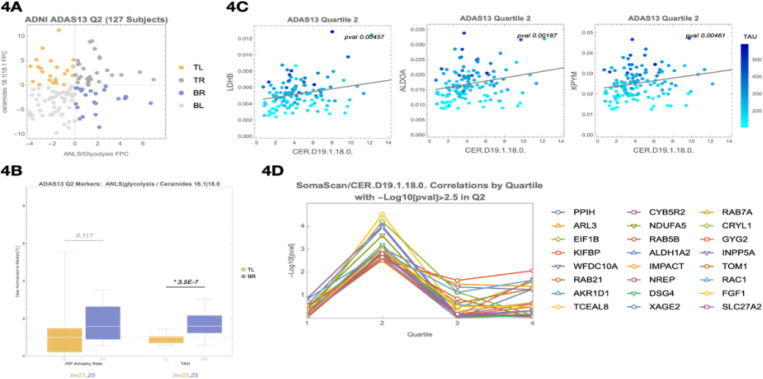
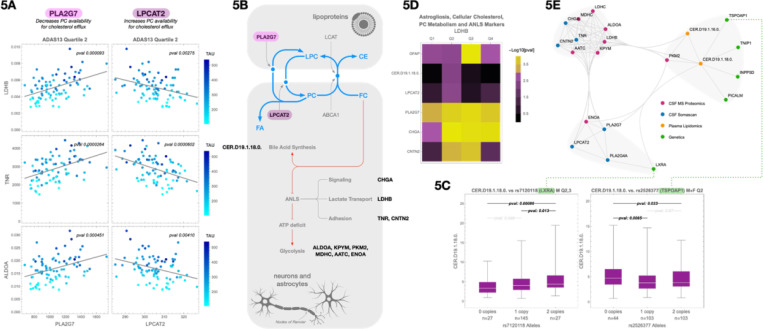
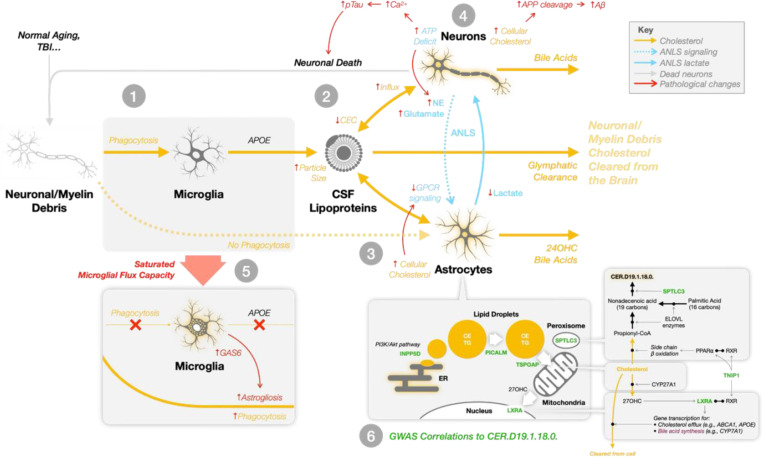
Age and APOE genotype are the strongest known risk factors for late-onset Alzheimer's disease (AD), but the mechanisms linking them to neuronal loss remain incompletely defined. Using multiomic data from the Alzheimer's Disease Neuroimaging Initiative (ADNI), we propose a unified hypothesis in which two interdependent failure modes-saturation of microglial lipid flux capacity and disruption of the astrocyte-neuron lactate shuttle (ANLS) due to excess astrocytic membrane cholesterol-drive disease progression upstream of amyloid and tau pathology. Stratifying participants by cognitive score quartiles, we find consistent associations linking impaired lipid clearance, metabolic stress, and genetic variants regulating cholesterol handling. These processes appear to reinforce each other, resulting in accelerating neurodegeneration. Our hypothesis reframes AD as a systems-level collapse in metabolic coordination, rather than a purely linear pathological cascade. These insights emerged during the development of digital twin models for personalized interventions, highlighting the power of systems approaches to reveal hidden drivers of neurodegeneration.
Figures
Similar articles
-
Understanding disease progression and improving Alzheimer's disease clinical trials: Recent highlights from the Alzheimer's Disease Neuroimaging Initiative.Alzheimers Dement. 2019 Jan;15(1):106-152. doi: 10.1016/j.jalz.2018.08.005. Epub 2018 Oct 13. Alzheimers Dement. 2019. PMID: 30321505 Review.
-
Modulation of the Astrocyte-Neuron Lactate Shuttle System contributes to Neuroprotective action of Fibroblast Growth Factor 21.Theranostics. 2020 Jul 9;10(18):8430-8445. doi: 10.7150/thno.44370. eCollection 2020. Theranostics. 2020. PMID: 32724479 Free PMC article.
-
Possible Neuropathology of Sleep Disturbance Linking to Alzheimer's Disease: Astrocytic and Microglial Roles.Front Cell Neurosci. 2022 Jun 9;16:875138. doi: 10.3389/fncel.2022.875138. eCollection 2022. Front Cell Neurosci. 2022. PMID: 35755779 Free PMC article. Review.
-
Early increase of CSF sTREM2 in Alzheimer's disease is associated with tau related-neurodegeneration but not with amyloid-β pathology.Mol Neurodegener. 2019 Jan 10;14(1):1. doi: 10.1186/s13024-018-0301-5. Mol Neurodegener. 2019. PMID: 30630532 Free PMC article.
-
Multiple Effect of APOE Genotype on Clinical and Neuroimaging Biomarkers Across Alzheimer's Disease Spectrum.Mol Neurobiol. 2016 Sep;53(7):4539-47. doi: 10.1007/s12035-015-9388-7. Epub 2015 Aug 23. Mol Neurobiol. 2016. PMID: 26298664
References
-
- Alzheimer A., Stelzmann R. A., Schnitzlein H. N. & Murtagh F. R. An english translation of alzheimer’s 1907 paper, “über eine eigenartige erkankung der hirnrinde.” Clin. Anat. 8, 429–431 (1995). - PubMed
-
- Romero-Molina C., Garretti F., Andrews S. J., Marcora E. & Goate A. M. Microglial efferocytosis: Diving into the Alzheimer’s disease gene pool. Neuron 110, 3513–3533 (2022). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous