

Diversity and longitudinal records: Genetic architecture of disease associations and polygenic risk in the Taiwanese Han population
- PMID: 40465716
- PMCID: PMC12136033
- DOI: 10.1126/sciadv.adt0539
Diversity and longitudinal records: Genetic architecture of disease associations and polygenic risk in the Taiwanese Han population
Abstract
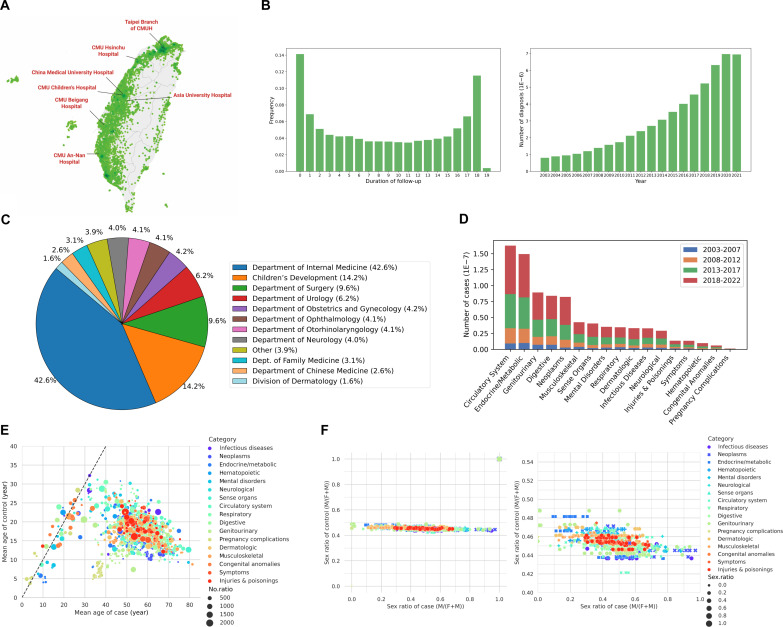
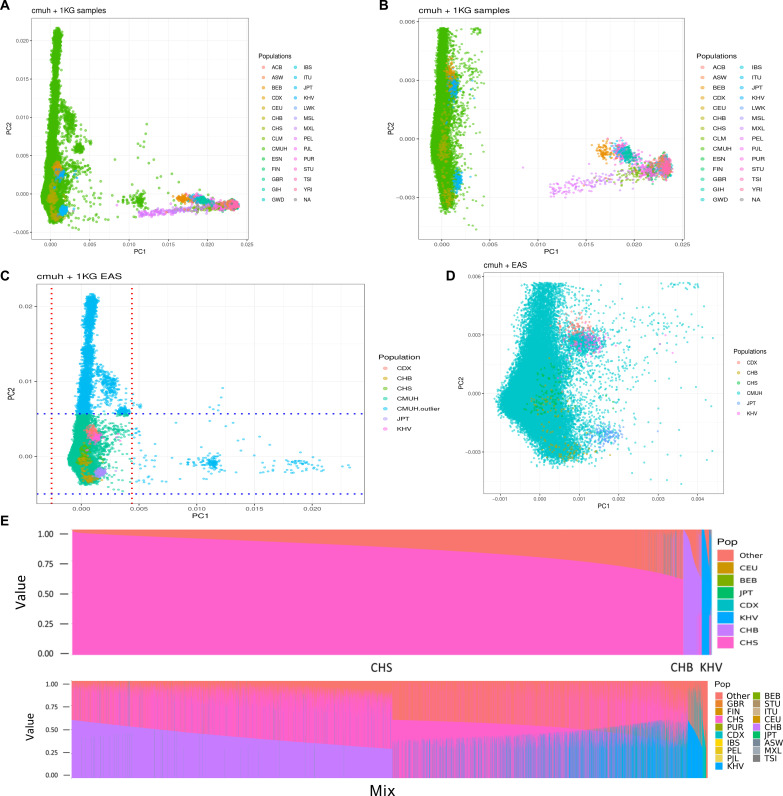
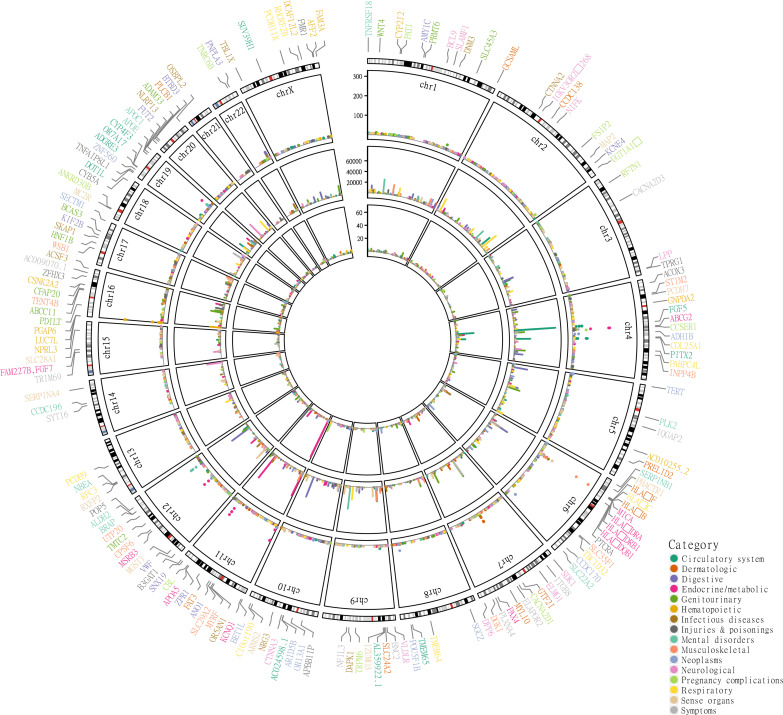
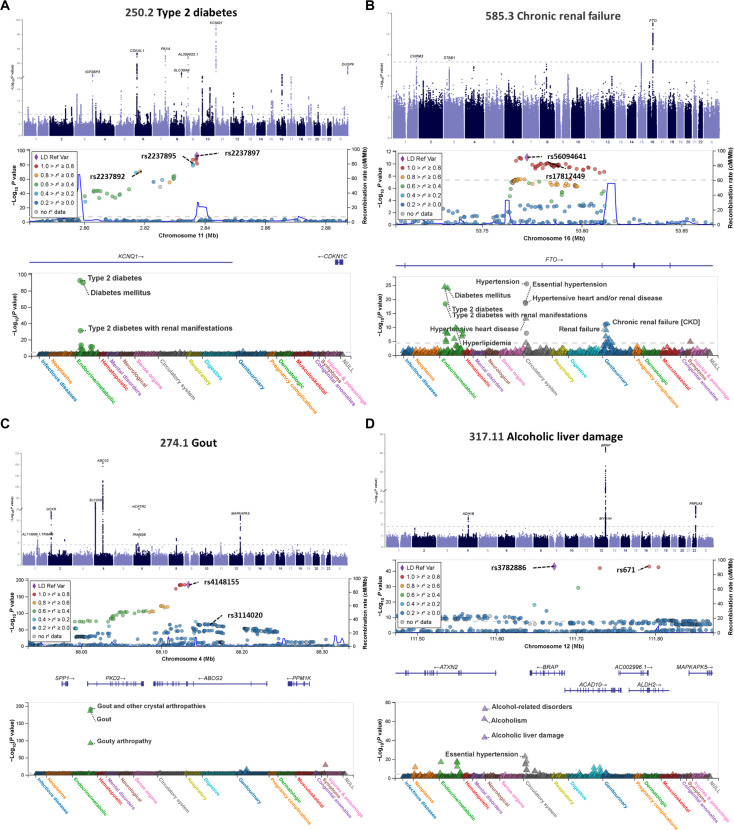
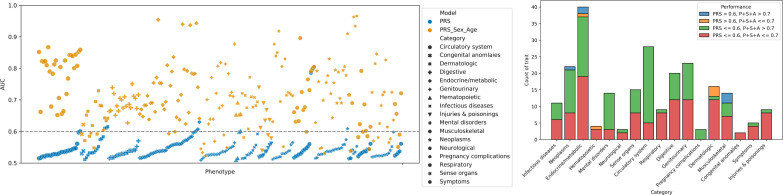
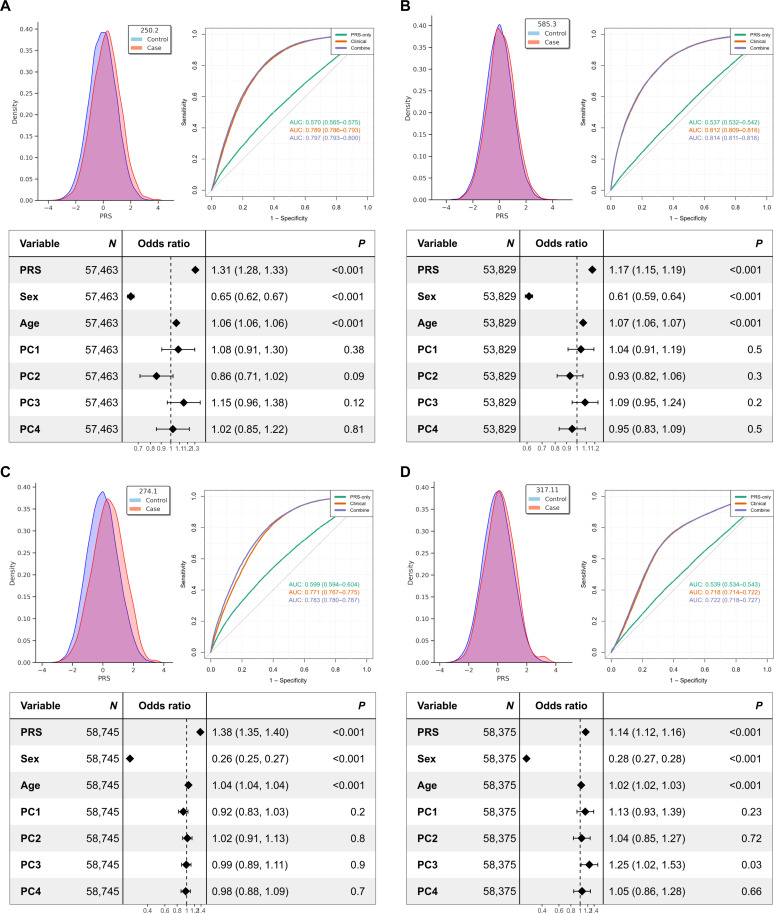
We addressed the underrepresentation of non-European populations in genome-wide association studies (GWASs) by building HiGenome, a large-scale genetic resource for the Taiwanese Han population. Using a custom genotyping array, we integrated deidentified electronic medical records (2003 to 2021) with genomic data to enable GWASs, phenome-wide association studies, and polygenic risk score (PRS) analysis. Among 413,000 participants, 323,397 passed ancestry and quality control filtering. GWASs covered 1085 traits, focusing on diseases prevalent in Taiwan such as type 2 diabetes, chronic kidney disease, gout, and alcoholic liver damage. PRSs were calculated for 238 traits, with the strongest associations observed in musculoskeletal disorders. Incorporating PRS into clinical practice supports early risk prediction and personalized prevention. To further expand translational value, we also conducted pharmacogenomic analysis and human leukocyte antigen typing. HiGenome offers a large-scale genetic and clinical dataset from the Taiwanese Han population, supporting population-specific analyses and precision medicine development in East Asia. The hospital-based design enables continuous follow-up and longitudinal data expansion.
Figures
References
-
- Huntley C., Torr B., Sud A., Rowlands C. F., Way R., Snape K., Hanson H., Swanton C., Broggio J., Lucassen A., Cartney M. M., Houlston R. S., Hingorani A. D., Jones M. E., Turnbull C., Utility of polygenic risk scores in UK cancer screening: A modelling analysis. Lancet Oncol. 24, 658–668 (2023). - PubMed
-
- Thomas S. A., Browning C. J., Charchar F. J., Klein B., Ory M. G., Bowden-Jones H., Chamberlain S. R., Transforming global approaches to chronic disease prevention and management across the lifespan: Integrating genomics, behavior change, and digital health solutions. Front. Public Health 11, 1248254 (2023). - PMC - PubMed
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
