

The RNA m6A landscape during human oocyte-to-embryo transition
- PMID: 40467862
- PMCID: PMC12264149
- DOI: 10.1038/s44318-025-00474-5
The RNA m6A landscape during human oocyte-to-embryo transition
Abstract
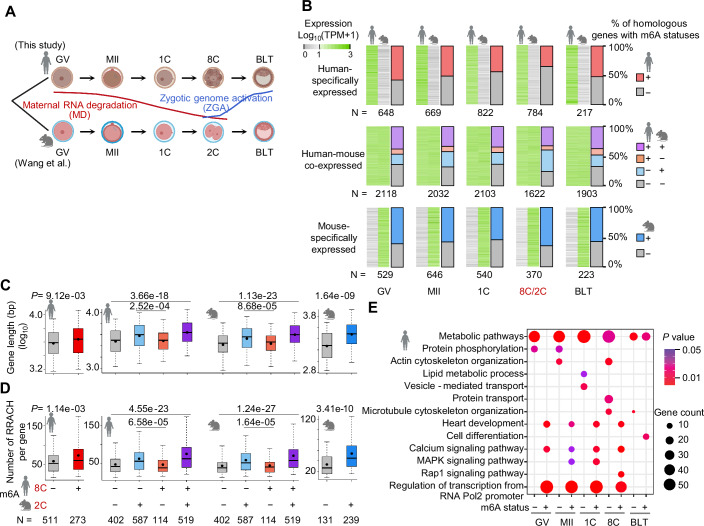
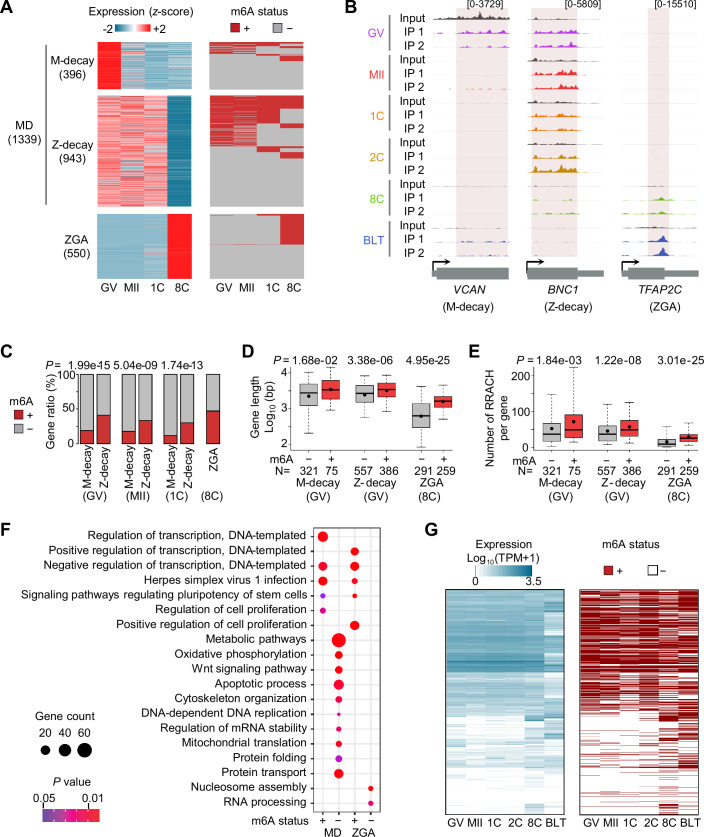
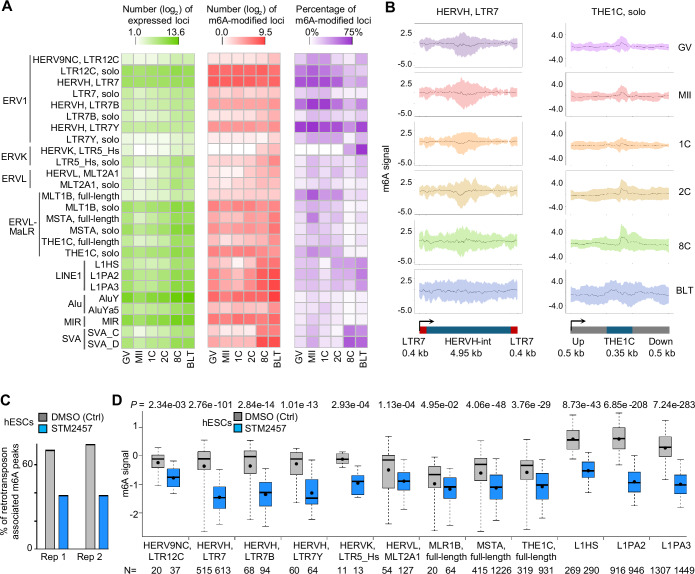
RNA N6-methyladenosine (m6A, m6A) modification is a critical regulator for a range of physiological processes. However, the dynamic m6A profiles within human preimplantation embryos remain uncharacterized. Here, we present the first RNA m6A landscape of single human oocytes and early embryos. Comparative analyses with mouse data reveal an intriguing divergence during the window of zygotic genome activation. m6A-modified genes are involved in regulation of gene transcription, while unmodified genes are mainly associated with basic metabolic processes. Maternal decay mRNAs exhibit a propensity for m6A modifications, and these genes are targeted by miRNAs. m6A modified genes that are constantly expressed across all stages demonstrate higher translation efficiency. Moreover, we observe frequent m6A enrichment on stage-specifically expressed retrotransposons, particularly within young subfamilies. m6A inhibitor leads to m6A erasure on massive retrotransposons. In summary, this study provides a resource to broaden our understanding about the regulatory roles of m6A during early human embryo development.
Keywords: Human Early Embryos; Retrotransposons; Zygotic Genome Activation; m6A.
© 2025. The Author(s).
Conflict of interest statement
Disclosure and competing interests statement. The authors declare no competing interests.
Figures










References
-
- Alberio R (2020) Regulation of cell fate decisions in early mammalian embryos. Annu Rev Anim Biosci 8:377–393 - PubMed
-
- Chelmicki T, Roger E, Teissandier A, Dura M, Bonneville L, Rucli S, Dossin F, Fouassier C, Lameiras S, Bourc’his D (2021) m(6)A RNA methylation regulates the fate of endogenous retroviruses. Nature 591:312–316 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
