

Molecular characterization of tick-borne bacterial and protozoan pathogens in parasitic ticks from Xinjiang, China
- PMID: 40468420
- PMCID: PMC12139197
- DOI: 10.1186/s13071-025-06857-1
Molecular characterization of tick-borne bacterial and protozoan pathogens in parasitic ticks from Xinjiang, China
Abstract
Background: Ticks are a type of hematophagous parasite, serving as critical vectors of pathogens that cause numerous human and animal diseases. Climate change has driven the geographical expansion of tick populations and increased the global transmission risk of tick-borne diseases. However, there has been a lack of comprehensive data on tick species distribution and their associated pathogen profiles in Xinjiang, China.
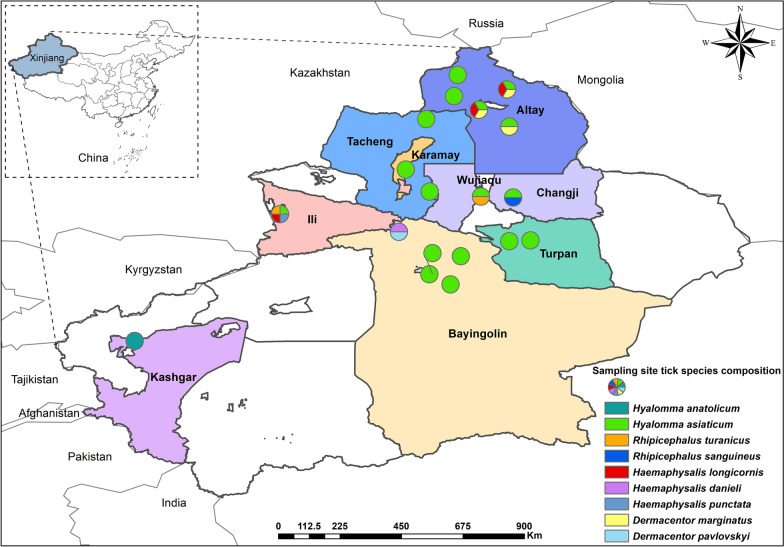
Methods: Ticks were collected from 19 sampling sites across nine regions in Xinjiang. The species were identified using both morphological and molecular biological methods. The presence of tick-borne bacterial and protozoan pathogens was detected through polymerase chain reaction (PCR). Finally, sequencing and phylogenetic analyses were performed to further characterize the identified ticks and pathogens.
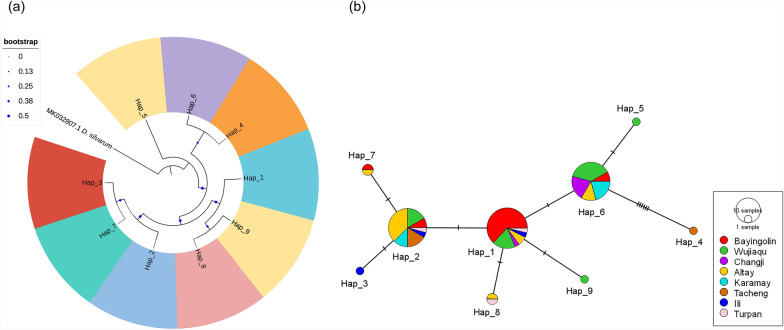
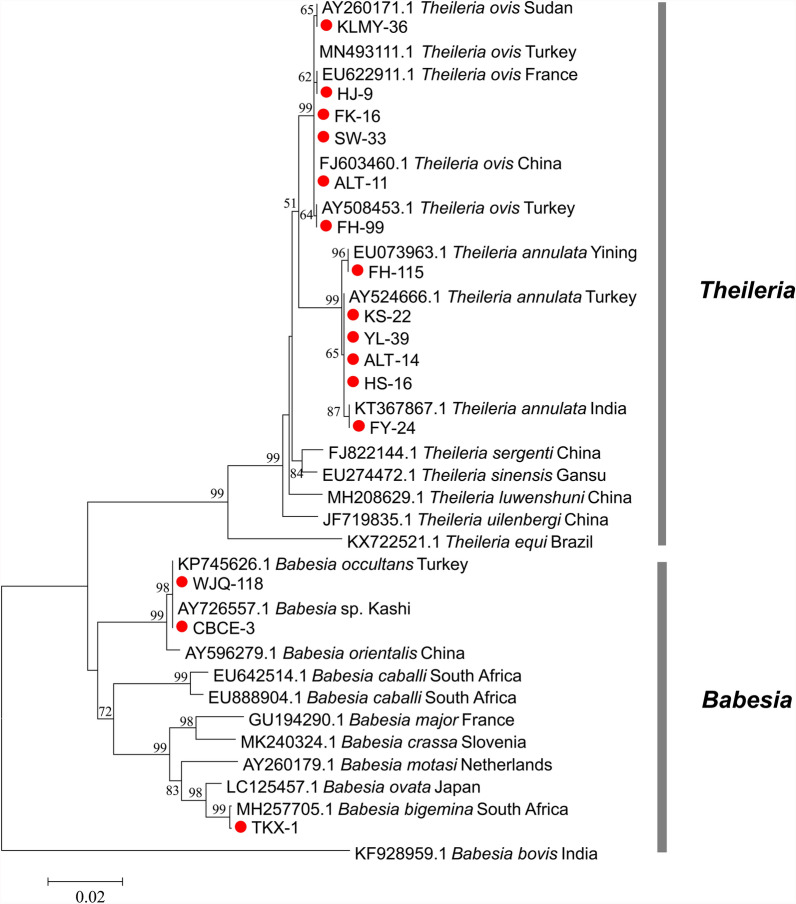
Results: A total of 1093 ticks were collected and identified, representing four genera and nine species, with Hyalomma asiaticum being the dominant species. Haplotype diversity and genetic differentiation analysis based on the 16S rRNA gene of the dominant species demonstrated that the Hy. asiaticum population in Xinjiang exhibits high haplotype diversity (Hd = 0.734), low nucleotide diversity (π = 0.00403), and significant genetic differentiation (Fst = 0.19716). Pathogen detection using PCR revealed an infection rate of 9.3% for Anaplasma, 18.1% for Rickettsia, and 9.0% for piroplasms. Phylogenetic analysis based on 16S rRNA sequences indicated that the Anaplasma genus identified in ticks comprised Anaplasma ovis, Anaplasma sp., and Anaplasma phagocytophilum. Phylogenetic analysis based on the opmA gene showed that the Rickettsia genus identified in ticks included Rickettsia aeschlimannii, Rickettsia conorii, Rickettsia slovaca, Rickettsia conorii subsp. raoultii, Rickettsia sp., Candidatus Rickettsia barbariae, and Candidatus Rickettsia jingxinensis. Similarly, phylogenetic analysis based on the 18S rRNA gene demonstrated that the piroplasms identified in ticks included Theileria annulata, Theileria ovis, Babesia bigemina, Babesia occultans, and Babesia sp. All gene sequences of the detected pathogens showed 99.8-100% identity with corresponding sequences deposited in GenBank.
Conclusions: This study demonstrates that Xinjiang harbors a rich diversity of tick species with a wide geographical distribution. Furthermore, the tick-borne pathogens in this region are complex and diverse. These results underscore the necessity of sustained and enhanced surveillance efforts targeting ticks and tick-borne diseases in this region.
Keywords: Anaplasma; Borrelia burgdorferi; Ehrlichia; Rickettsia; Piroplasm; Tick species; Tick-borne pathogens; Xinjiang.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: This study was approved by the Scientific Ethics Committee of Huazhong Agricultural University. Written informed consent was obtained from herders and farm owners prior to sampling. All procedures strictly adhered to the noninvasive principle, ensuring that host animals (cattle and sheep) experienced no additional harm or stress during the process. Ticks were collected solely through superficial examination and mechanical removal from the epidermis of the animal. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures



References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
