

Single-Cell and Spatial Transcriptomic Profiling of Penile Squamous Cell Carcinoma Reveals Dynamics of Tumor Differentiation and Immune Microenvironment
- PMID: 40470730
- PMCID: PMC12412502
- DOI: 10.1002/advs.202500216
Single-Cell and Spatial Transcriptomic Profiling of Penile Squamous Cell Carcinoma Reveals Dynamics of Tumor Differentiation and Immune Microenvironment
Abstract
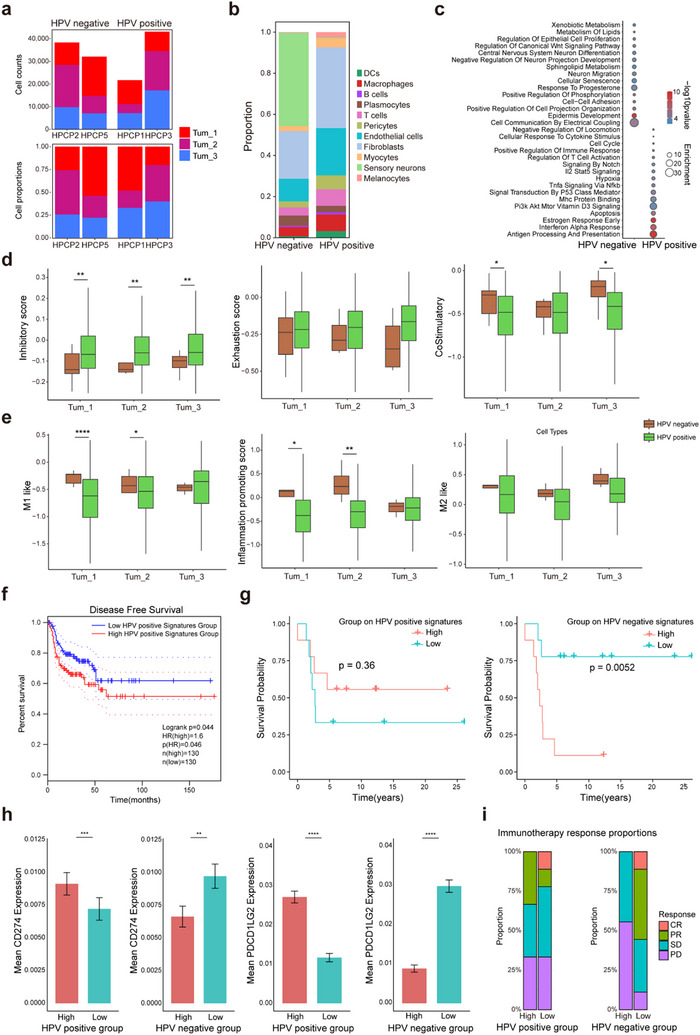
Penile squamous cell carcinoma (PSCC) is a highly aggressive malignancy without effective treatment due to limited knowledge of its development and tumor microenvironment (TME). In this study, single-nucleus RNA sequencing (snRNA-seq) and high-resolution spatial transcriptomics are employed to comprehensively investigate the development trajectories and the TME. The results revealed that PSCC cells mimicked the differentiation and tissue organization of normal penile epithelium, independent of the human papillomavirus (HPV) infection status. Notably, a spatial subtype, Tum_1, appeared at early stage of tumor differentiation and in tumor-normal boundary regions. This subtype exhibited enhanced basal-like and stemness features and showed high LAMC2 expression, which activated laminin-integrin signaling via ITGA6/ITGB4, promoting tumor invasiveness. Furthermore, the results indicated that HPV-positive basal stem-like neoplasms dampened the immune function of T cells and macrophages, promoting an immunosuppressive environment that facilitates tumor progression. Supporting this, the patients with head and neck squamous cell carcinoma and lung squamous cell carcinoma who have high expression of HPV-positive Tum_1 signatures derived greater benefit from PD-1 blockade therapy. In summary, the findings provide a comprehensive spatial landscape of the PSCC TME and suggest potential treatment approaches targeting laminin-integrin interaction and immunotherapy, especially in HPV-positive patients.
Keywords: penile squamous cell carcinoma; single‐nucleus RNA sequencing; spatial landscape; spatial transcriptomics; tumor microenvironment.
© 2025 The Author(s). Advanced Science published by Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
-
- Thomas A., Necchi A., Muneer A., Tobias‐Machado M., Tran A. T. H., Van Rompuy A. S., Spiess P. E., Albersen M., Nat. Rev. Dis. Primers 2021, 7, 11. - PubMed
-
- Bray F., Laversanne M., Sung H., Ferlay J., Siegel R. L., Soerjomataram I., Jemal A., CA Cancer J. Clin. 2024, 74, 229. - PubMed
-
- Brouwer O. R., Rumble R. B., Ayres B., Sanchez Martinez D. F., Oliveira P., Spiess P. E., Johnstone P. A. S., Crook J., Pettaway C. A., Tagawa S. T., JCO Oncol. Pract. 2024, 20, 33. - PubMed
-
- Elst L., Vreeburg M., Brouwer O., Albersen M., Eur. Urol. Focus 2023, 9, 241. - PubMed
-
- Brouwer O. R., Albersen M., Parnham A., Protzel C., Pettaway C. A., Ayres B., Antunes‐Lopes T., Barreto L., Campi R., Crook J., Fernandez‐Pello S., Greco I., van der Heijden M. S., Johnstone P. A. S., Kailavasan M., Manzie K., Marcus J. D., Necchi A., Oliveira P., Osborne J., Pagliaro L. C., Garcia‐Perdomo H. A., Rumble R. B., Sachdeva A., Sakalis V. I., Zapala L., Sanchez Martinez D. F., Spiess P. E., Tagawa S. T., Eur. Urol. 2023, 83, 548. - PubMed
MeSH terms
Grants and funding
- 2021YFB3801000/National Key Research and Development Program of China
- 2022YFA1103400/National Key Research and Development Program of China
- 82171999/National Natural Science Foundation of China
- 82302266/National Natural Science Foundation of China
- 82402416/National Natural Science Foundation of China
- 31021240077/Talent Support Program of Harbin Medical University
- CYQN24038/Chun-Yan Innovation Team Program from Heilongjiang Provincial Department of Science and Technology
- Nn102024-01/Nn10 project at Harbin Medical University Cancer Hospital
- 31021240027/"Young Marshal Unveiling" Project of Harbin Medical University
- XJ2024052/Hong Kong scholar project
- YQ2024H023/Natural Science Foundation of Heilongjiang Province
- YQJH2023034/Key Project of Heilongjiang Province Outstanding Young Teachers Basic Research
- LBH-Z22030/Heilongjiang Provincial Department of Human Resources and Social Security
- 2021M693828/Heilongjiang Provincial Postdoctoral Science Foundation
- JJYQ2024-03/Harbin Medical University Cancer Hospital Haiyan Foundation
- JJZD2024-05/Harbin Medical University Cancer Hospital Haiyan Foundation
- JJZD2024-24/Harbin Medical University Cancer Hospital Haiyan Foundation
- 2022ZXJ03C07/Key Research and Development Program of Heilongjiang Province
- 2022ZX06004/Key Research and Development Program of Heilongjiang Province
LinkOut - more resources
Full Text Sources
Miscellaneous