

A phase I/II trial of WT1-specific TCR gene therapy for patients with acute myeloid leukemia and active disease post-allogeneic hematopoietic cell transplantation: skewing towards NK-like phenotype impairs T cell function and persistence
- PMID: 40473616
- PMCID: PMC12141728
- DOI: 10.1038/s41467-025-60394-0
A phase I/II trial of WT1-specific TCR gene therapy for patients with acute myeloid leukemia and active disease post-allogeneic hematopoietic cell transplantation: skewing towards NK-like phenotype impairs T cell function and persistence
Abstract
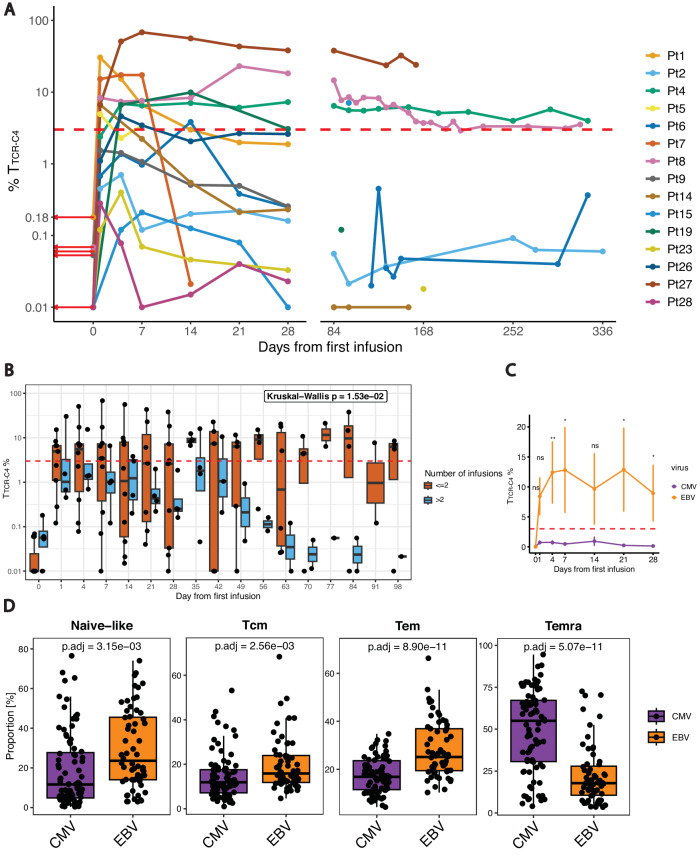
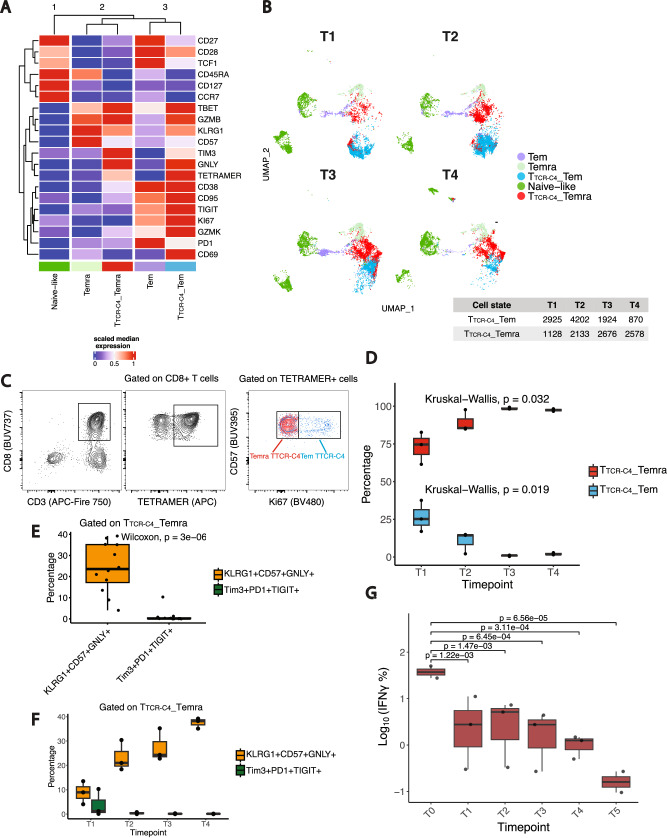
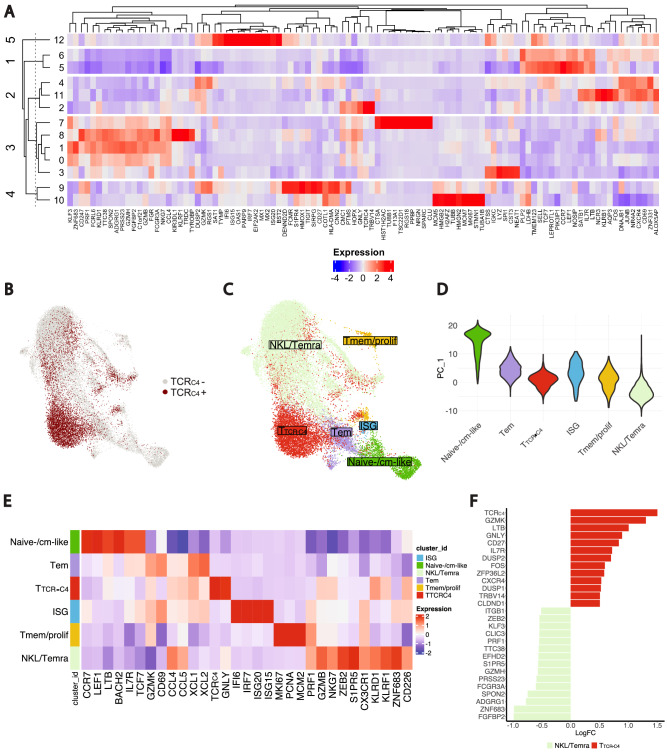
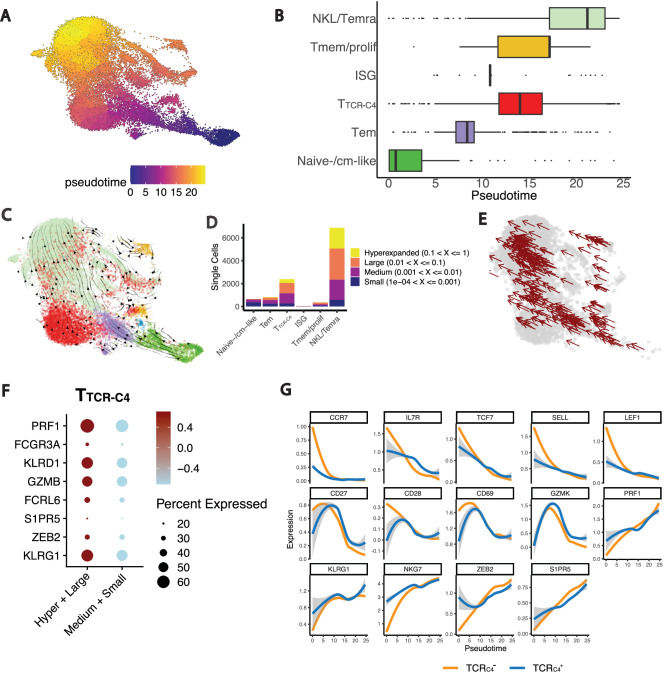
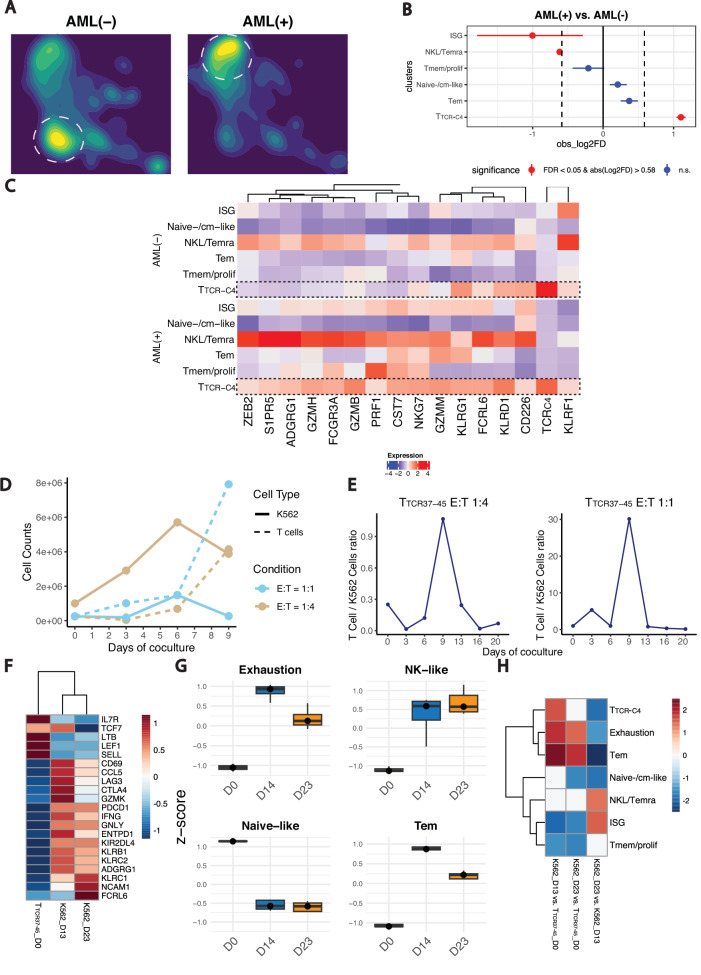
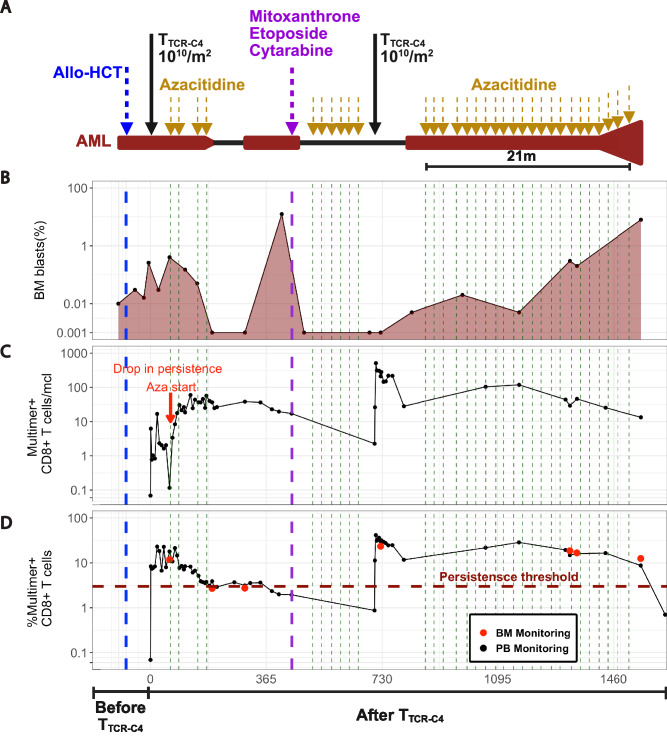
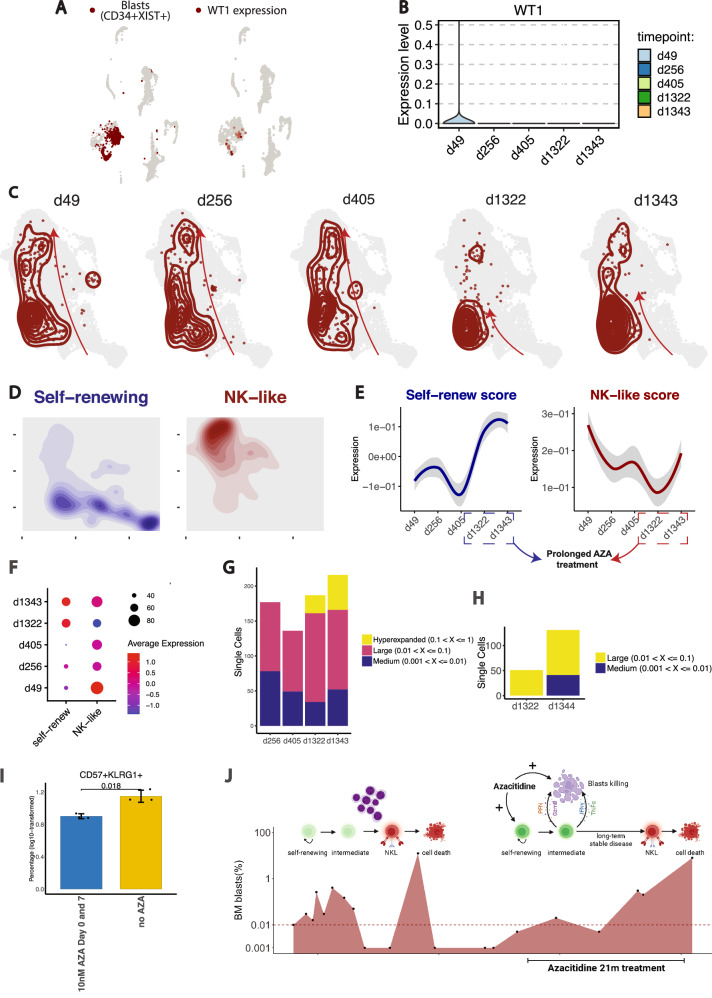
Relapsed and/or refractory acute myeloid leukemia (AML) post-allogeneic hematopoietic cell transplantation (HCT) is usually fatal. We previously reported that post-HCT immunotherapy with Epstein-Barr virus (EBV)-specific donor CD8+ T cells engineered to express a Wilms Tumor Antigen 1-specific T-cell receptor (TTCR-C4) appeared to prevent relapse in high-risk patients. In this phase I/II clinical trial (NCT01640301), we evaluated safety (primary endpoint), persistence and efficacy (secondary endpoints) of EBV- or Cytomegalovirus (CMV)-specific TTCR-C4 in fifteen patients with active AML post-HCT. Infusions were well tolerated, with no dose-limiting toxicities or serious adverse events related to the product. However, TTCR-C4 cells did not clearly improve outcomes despite EBV-specific TTCR-C4 cells showing enhanced potential for prolonged persistence compared to CMV-specific TTCR-C4. Investigating the fate of persisting TTCR-C4, we identified a shift towards natural killer-like (NKL) terminal differentiation, distinct from solid tumor-associated canonical exhaustion programs. In one patient, treatment with azacitidine appeared to mitigate this NKL skewing, promoting TTCR-C4 persistence. These findings suggest that AML drives a distinct form of T-cell dysfunction, highlight the need for targeted approaches that preserve T-cell fitness, ultimately improving the efficacy of cellular therapies for AML.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: A.G.C. has received support from Juno Therapeutics, Lonza, and Affini-T. P.D.G. is a consultant, has received support from and has had ownership interest in Juno Therapeutics and Affini-T Therapeutics. He has also received support from Lonza, and consults and has ownership interest in Rapt Therapeutics, Elpiscience, Immunoscape, Earli, Metagenomi. Catalia, and Nextech. P.D.G., T.M.S., and the Fred Hutchinson Cancer Research Center have intellectual property related to TCRC4. A.G.C. and K.G.P. have received reagents from 10X Genomics. R.G. has received consulting income from Takeda, Arcellx and Sanofi, and declares ownership in Ozette Technologies. CJW receives equity from BionTech, and is a SAB member of Repertoire, Adventris, Aethon Therapeutics and Nature’s Toolbox. M.B. is employed at Bristol Myers Squibb. The remaining authors declare no competing interests.
Figures
References
-
- Schmid, C. et al. Treatment, risk factors, and outcome of adults with relapsed AML after reduced intensity conditioning for allogeneic stem cell transplantation. Blood119, 1599–1606 (2012). - PubMed
-
- Haque, T. et al. Complete regression of posttransplant lymphoproliferative disease using partially HLA-matched Epstein Barr virus-specific cytotoxic T cells. Transplantation72, 1399–1402 (2001). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- n/a/Fred Hutchinson Cancer Research Center (Hutchinson Center)
- P01 CA018029/CA/NCI NIH HHS/United States
- 5K08CA169485/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- K08 CA169485/CA/NCI NIH HHS/United States
- n/a/Damon Runyon Cancer Research Foundation (Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation)
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
