

Comprehensive analysis of Saccharomyces cerevisiae intron structures in vivo
- PMID: 40473812
- PMCID: PMC12350175
- DOI: 10.1038/s41594-025-01565-x
Comprehensive analysis of Saccharomyces cerevisiae intron structures in vivo
Abstract
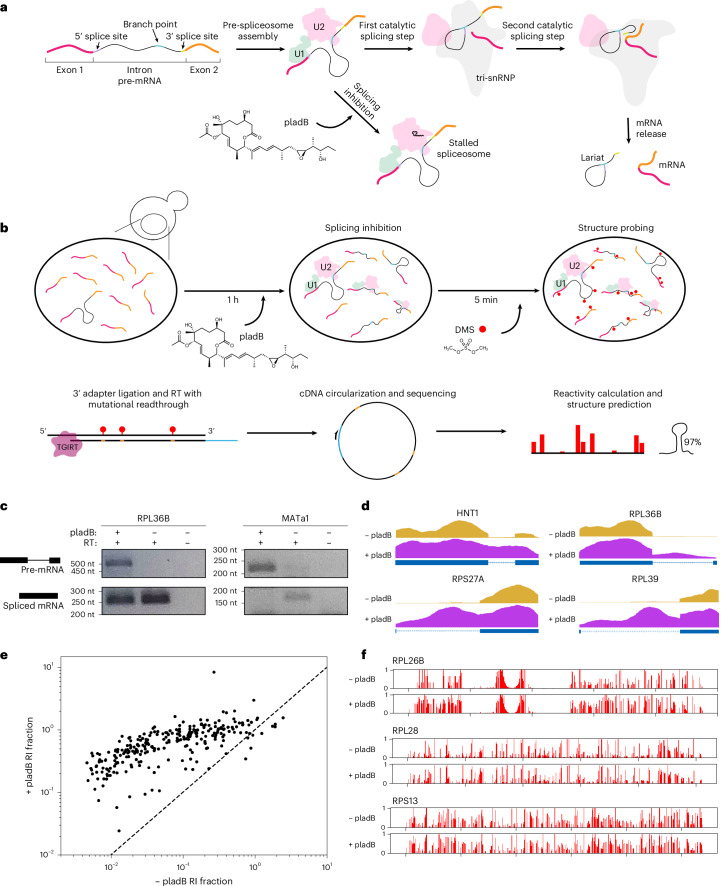
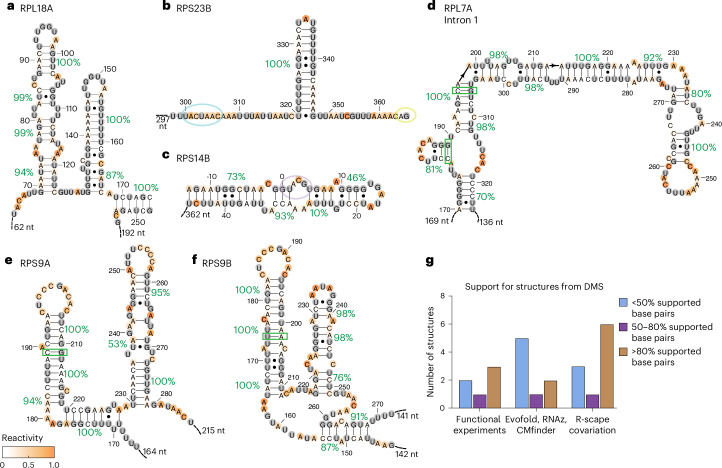
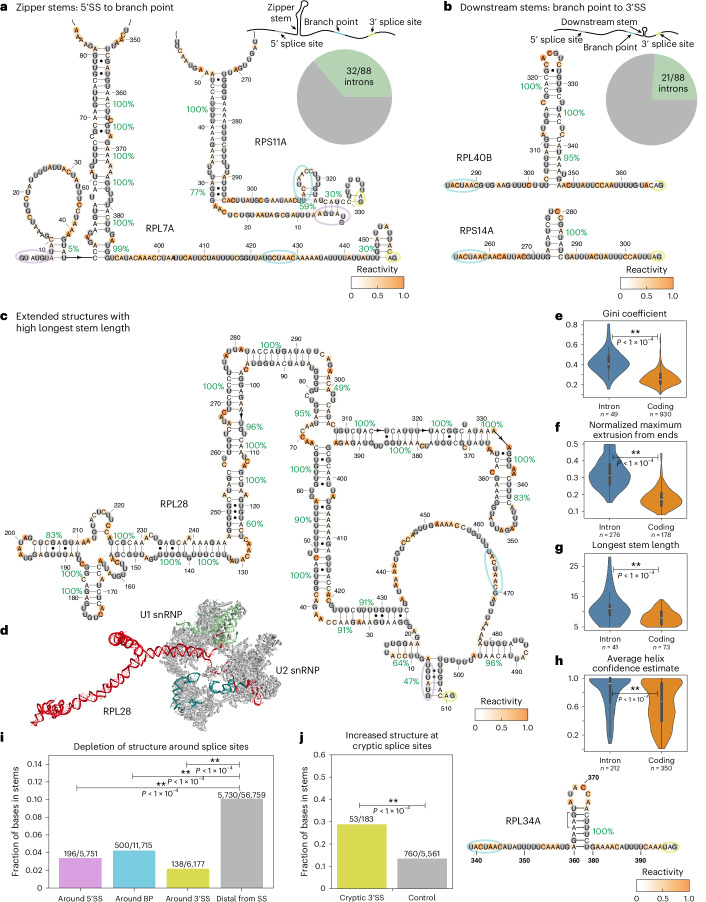
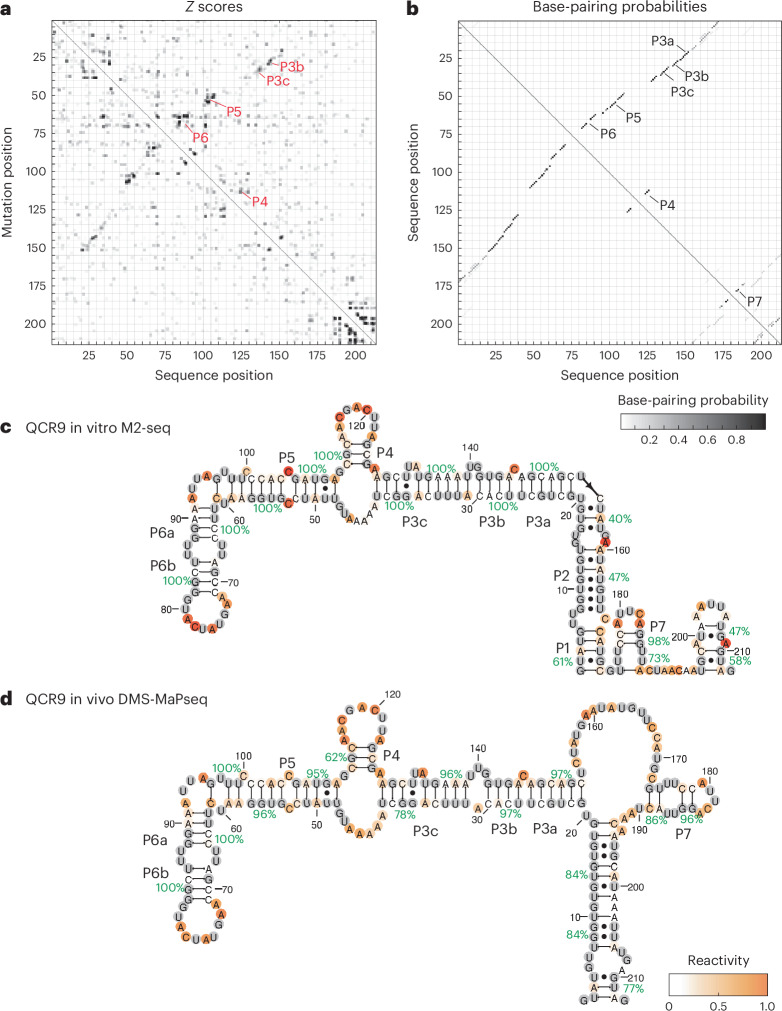
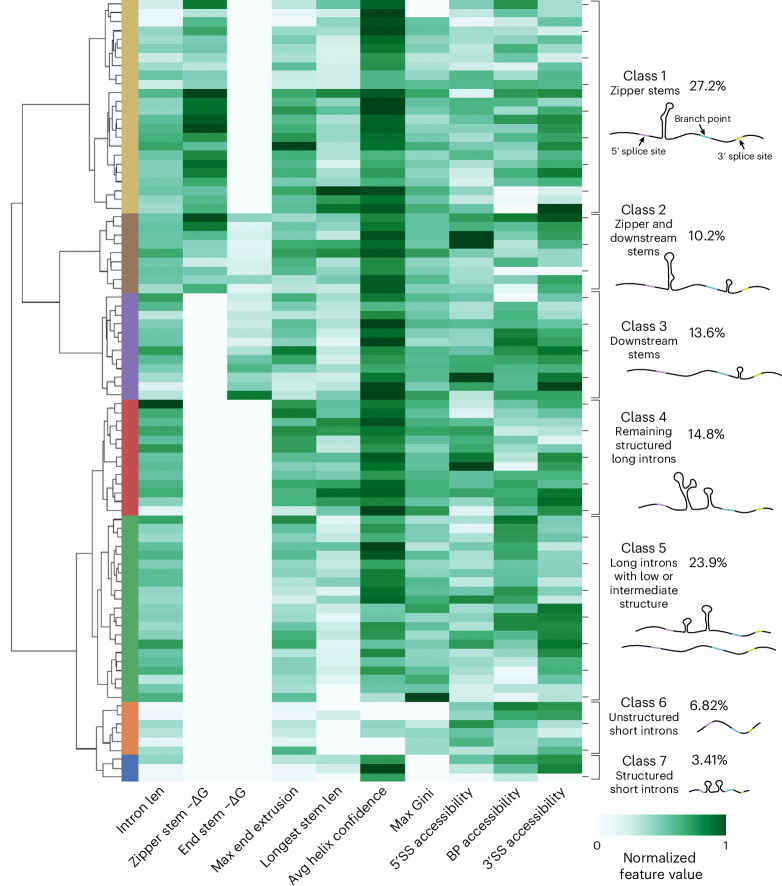
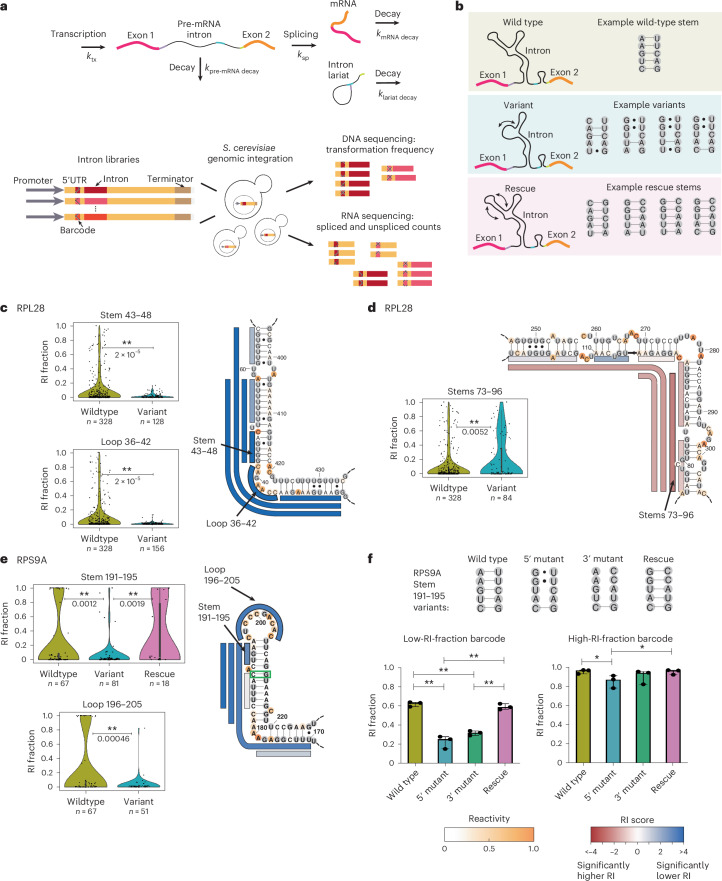
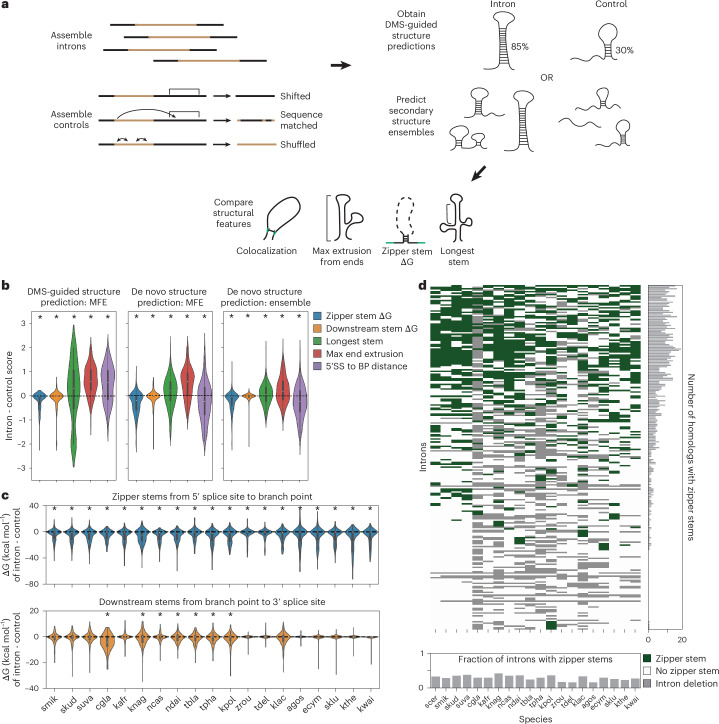
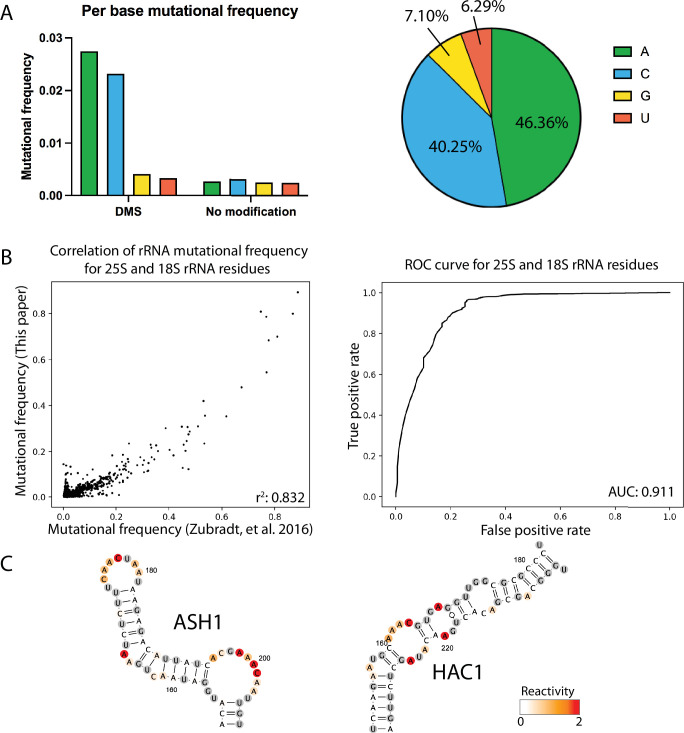
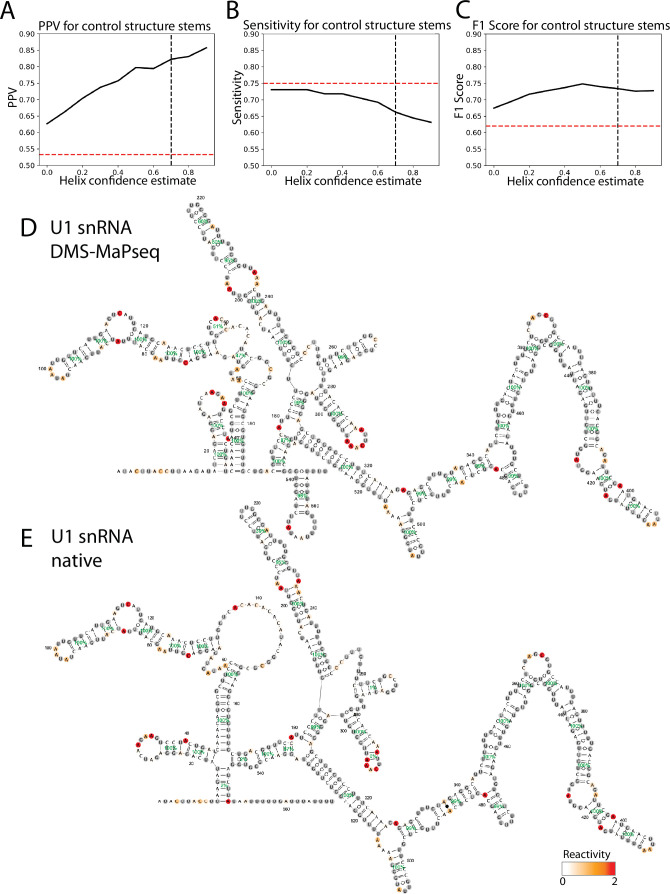
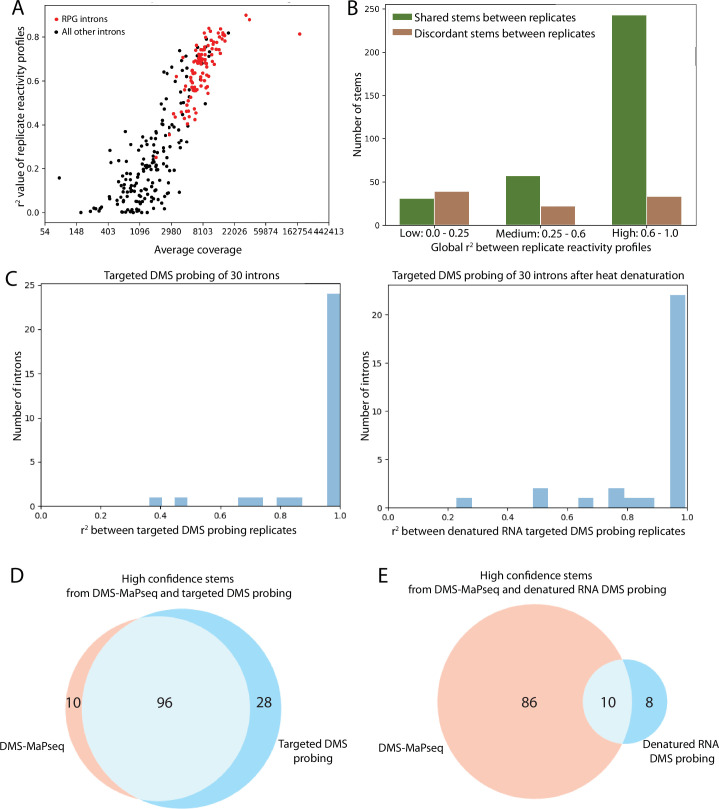
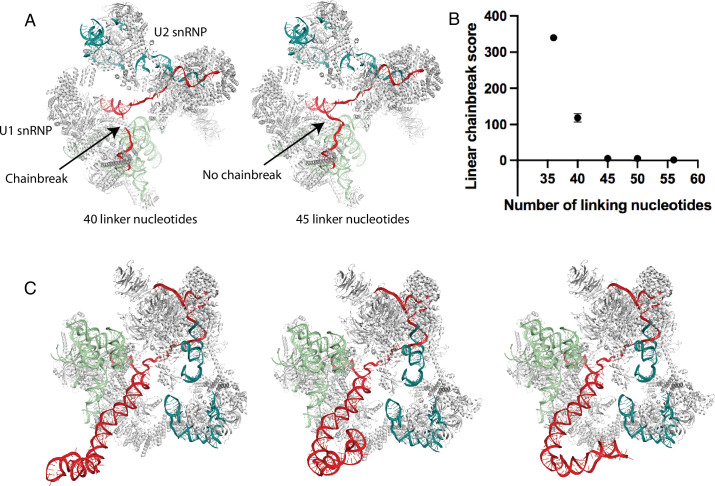
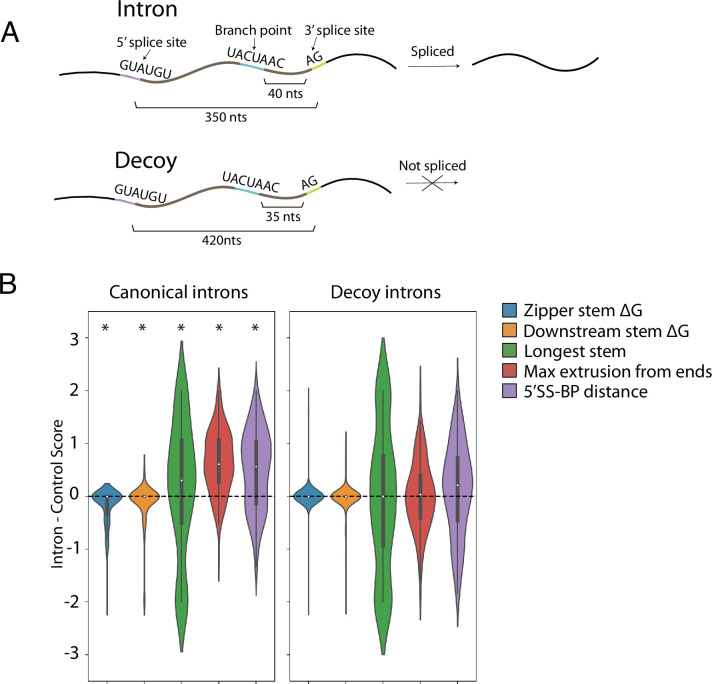
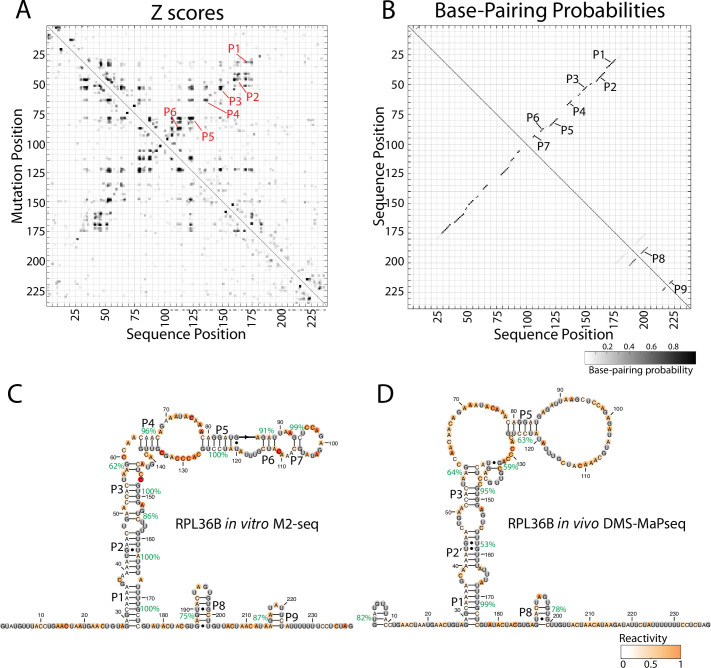
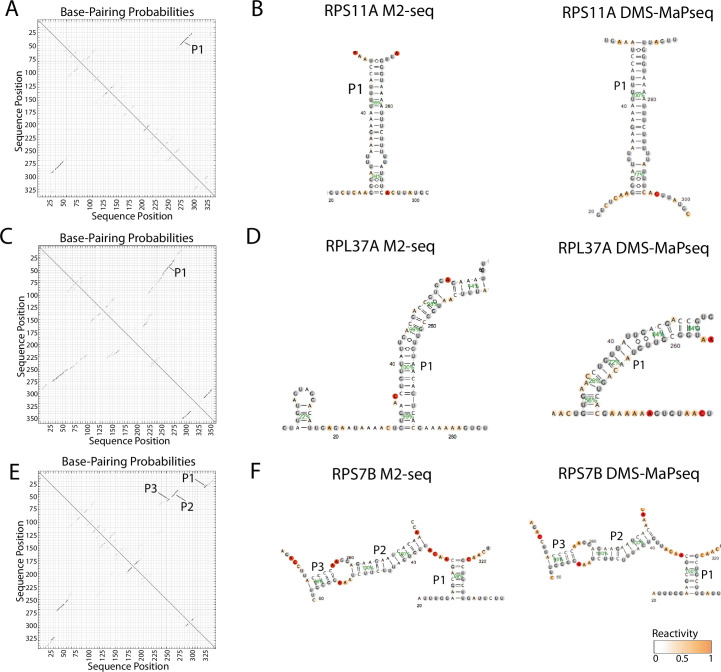
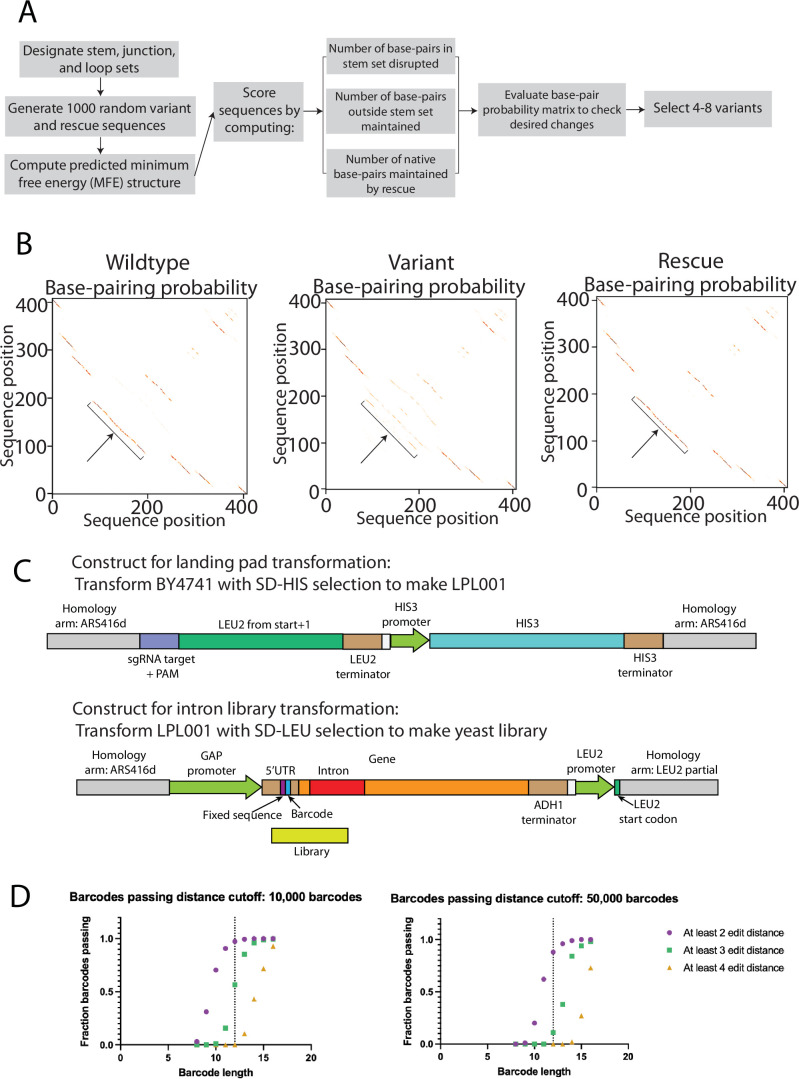
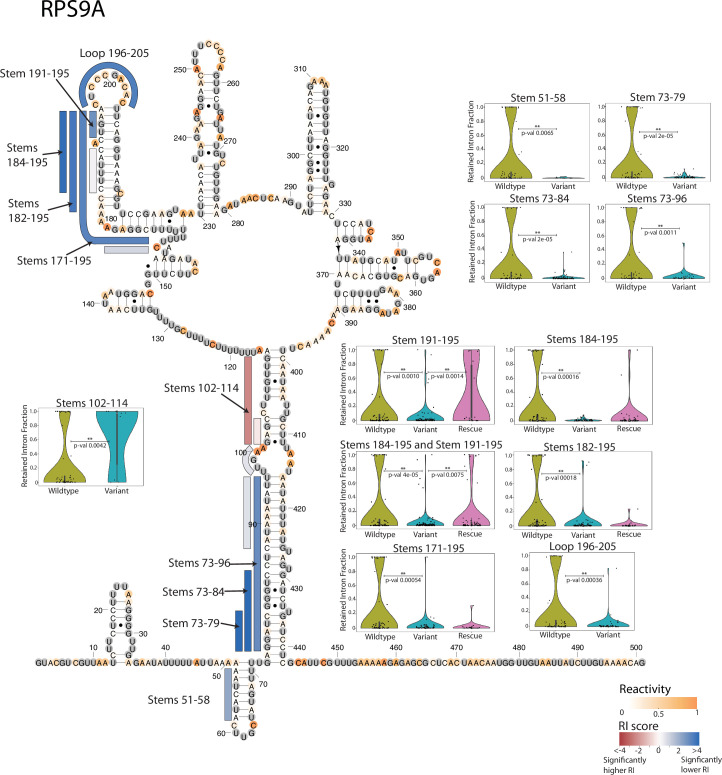
Pre-mRNA secondary structures are hypothesized to regulate RNA processing pathways, but such structures have been difficult to visualize in vivo. Here, we characterize Saccharomyces cerevisiae pre-mRNA structures through transcriptome-wide dimethyl sulfate probing, enriching for low-abundance pre-mRNA through splicing inhibition. We cross-validate structures found from phylogenetic and mutational studies and identify structures within the majority of measured introns (79 of 88). We find widespread formation of 'zipper stems' between the 5' splice site and branch point, 'downstream stems' between the branch point and the 3' splice site, and previously uncharacterized long stems that distinguish pre-mRNA from spliced mRNA. Multi-dimensional chemical mapping reveals intron structures that independently form in vitro without the presence of binding partners, and structure ensemble prediction suggests that such structures appear in introns across the Saccharomyces genus. We further develop a high-throughput functional assay to characterize variants of RNA structure (VARS-seq), applying it to 135 sets of stems across 7 introns, identifying structured elements that alter retained intron levels at a distance from canonical splice sites. This transcriptome-wide inference of intron RNA structures introduces alternative paradigms and model systems for understanding how pre-mRNA folding influences gene expression.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
Cells resist starvation through a nutrient stress splice switch.Nucleic Acids Res. 2025 Jun 20;53(12):gkaf525. doi: 10.1093/nar/gkaf525. Nucleic Acids Res. 2025. PMID: 40550515 Free PMC article.
-
Survivor, family and professional experiences of psychosocial interventions for sexual abuse and violence: a qualitative evidence synthesis.Cochrane Database Syst Rev. 2022 Oct 4;10(10):CD013648. doi: 10.1002/14651858.CD013648.pub2. Cochrane Database Syst Rev. 2022. PMID: 36194890 Free PMC article.
-
Surgery for epilepsy.Cochrane Database Syst Rev. 2015 Jul 1;(7):CD010541. doi: 10.1002/14651858.CD010541.pub2. Cochrane Database Syst Rev. 2015. Update in: Cochrane Database Syst Rev. 2019 Jun 25;6:CD010541. doi: 10.1002/14651858.CD010541.pub3. PMID: 26130264 Updated.
Cited by
-
Autoregulation of RPL7B by inhibition of a structural splicing enhancer.Nucleic Acids Res. 2025 Jul 19;53(14):gkaf739. doi: 10.1093/nar/gkaf739. Nucleic Acids Res. 2025. PMID: 40744491 Free PMC article.
References
-
- Wilkinson, M. E., Charenton, C. & Nagai, K. RNA splicing by the spliceosome. Annu. Rev. Biochem.89, 359–388 (2020). - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
