

Mutual suppression between mutations in the Dictyostelium Greenbeard pathway restores wild-type development
- PMID: 40474076
- PMCID: PMC12142906
- DOI: 10.1186/s12864-025-11745-0
Mutual suppression between mutations in the Dictyostelium Greenbeard pathway restores wild-type development
Abstract
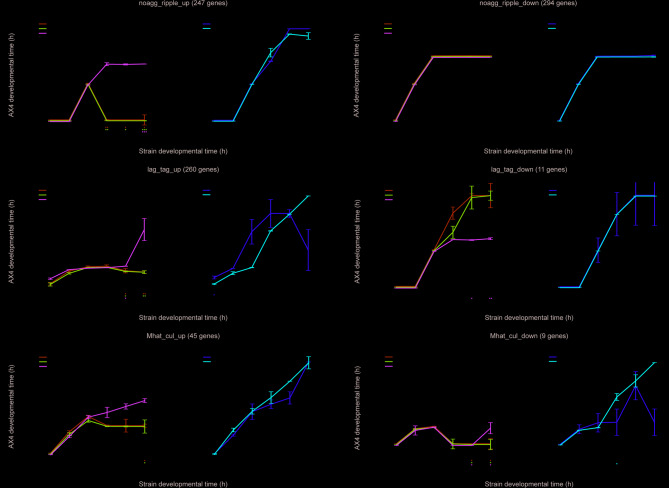
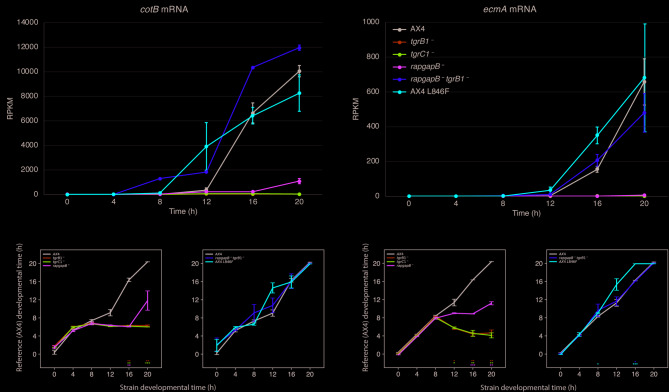
Background: The Dictyostelium greenbeard pathway is mediated by two polymorphic transmembrane proteins, the TgrC1 ligand and the TgrB1 receptor. These proteins mediate allorecognition, altruism, and the developmental transition to multicellularity. A genetic suppressor screen revealed activating mutations in tgrB1 and inactivating mutations in rapgapB, a regulator of the GTPase protein RapA. Inactivation of either tgrB1, tgrC1, or rapgapB leads to developmental defects, but the respective double-mutant strains rapgapB-tgrB1- and rapgapB-tgrC1- develop well and produce spores. This mutual suppression could result from inducing an alternative pathway or from restoring wild-type development, but morphological analyses alone could not resolve this question.
Results: Here, we show that the mutual suppression between rapgapB- and tgrB1- restores wild-type development. We also analyzed an activated tgrB1 allele in the wild-type background and found evidence for interactions between the wild-type and the activated alleles. Using RNA-sequencing analyses, we compared the transcriptomes of the wild type to those of several mutant strains and found that the single-gene mutations attenuated transcriptome progression over developmental time, whereas the double-gene mutation strain rapgapB-tgrB1- and the activated tgrB1 mutation exhibited near wild-type transcriptomes. Our findings suggest that tgrB1, tgrC1, and rapgapB are involved in a pathway in which rapgapB negatively regulates tgrB1 and tgrC1 expression, whereas tgrB1 and tgrC1 positively regulate rapgapB expression.
Conclusions: These findings suggest that the Dictyostelium greenbeard pathway interfaces with the central RapGAPB-RapA regulatory pathway, providing molecular insight into a mutual suppression mechanism in which two deleterious mutations restore wild-type behavior.
Keywords: Dictyostelium allorecognition; Dictyostelium development; Greenbeard pathway; Mutual suppression; Transcriptome phenotyping.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures



References
-
- Kessin RH. Dictyostelium - Evolution, cell biology, and the development of multicellularity. Cambridge, UK: Cambridge Univ. Press; 2001.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
