

Disseminated intravascular coagulation
- PMID: 40474215
- PMCID: PMC12143096
- DOI: 10.1186/s40560-025-00794-y
Disseminated intravascular coagulation
Abstract
Background: Disseminated intravascular coagulation (DIC) is characterized by systemic coagulation activation, anticoagulation pathway impairment, and persistent fibrinolysis suppression, resulting in widespread microvascular thrombosis, followed by hemorrhagic consumption coagulopathy and multiple organ dysfunction syndrome. This article aimed to provide a comprehensive and updated DIC overview.
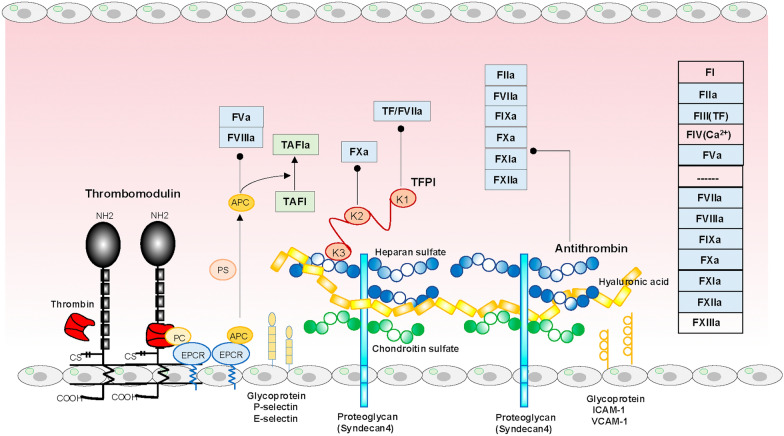
Main body: The International Society on Thrombosis and Hemostasis provides definitions, underlying disorders, diagnostic algorithms, and management guidelines for DIC. Two clinical features of DIC are hemorrhagic consumption coagulopathy, characterized by oozing and difficult-to-control bleeding, and microvascular thrombosis, leading to dysfunctions in multiple vital organs. Histones derived from cellular damage play central roles in the innate-immune-based coagulation model, comprising the initiation, amplification, propagation, and reinforcement phases, which, if dysregulated, develop into DIC. Thus, the innate immune-mediated pathogenic pathways in DIC have become clear. Cell death, damage-associated molecular patterns (including histones), crosstalk between hypoxic inflammation and coagulation, and the serine protease network (comprising coagulation and fibrinolysis, the Kallikrein-Kinin system, and complement pathways) play major roles in DIC pathogenesis. Conversely, these pathogenic pathways and DIC synergistically contribute to organ dysfunction, leading to poor prognoses. Effective DIC management requires treating the underlying condition, along with substitution therapies and, in some cases, antifibrinolytics. Anticoagulant use has been extensively debated; however, the selection of optimal target patients could optimize their application and improve patient outcomes in the near future.
Conclusions: This review provides an updated overview of DIC, aiming to help readers understand various aspects of DIC today.
Keywords: Cell death; Complement; Disseminated intravascular coagulation (DIC); Hemorrhage; Histone; Hypoxia; Inflammation; Innate immunity; Management; Organ dysfunction.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures






References
-
- Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primers. 2016;2:16037. - PubMed
-
- Colman RW, Robboy SJ, Minna JD. Disseminated intravascular coagulation (DIC): an approach. Am J Med. 1972;52:679–89. - PubMed
-
- Esmon CT, Fukudome K, Mather T, Bode W, Regan LM, Stearns-Kurosawa DJ, et al. Inflammation, sepsis, and coagulation. Haematologica. 1999;84:254–9. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
