

This is a preprint.
Partitioned Multi-MUM finding for scalable pangenomics
- PMID: 40475428
- PMCID: PMC12139944
- DOI: 10.1101/2025.05.20.654611
Partitioned Multi-MUM finding for scalable pangenomics
Update in
-
Partitioned multi-MUM finding for scalable pangenomics with MumemtoM.Genome Res. 2026 Feb 3;36(2):397-404. doi: 10.1101/gr.280940.125. Genome Res. 2026. PMID: 41203424
Abstract
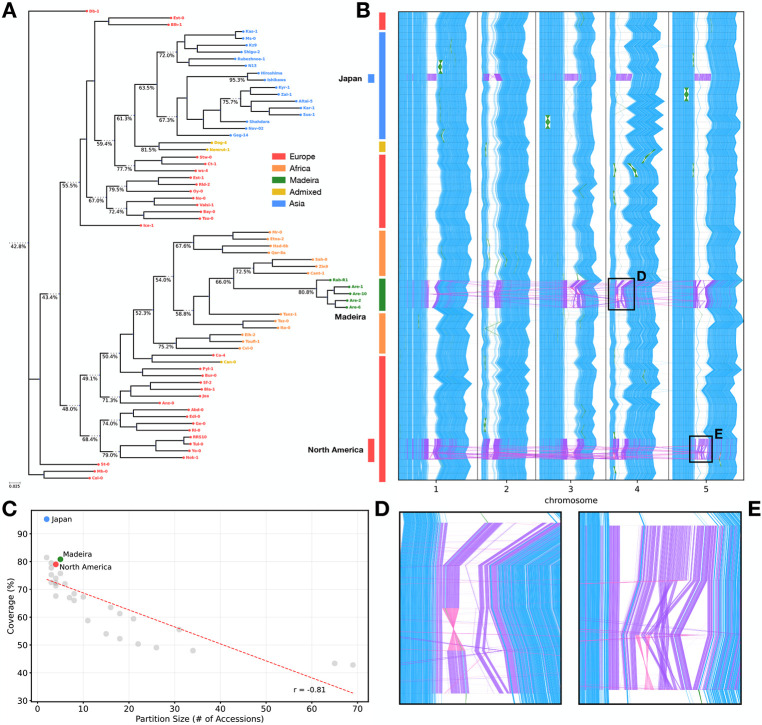
Pangenome collections are growing to hundreds of high-quality genomes. This necessitates scalable methods for constructing pangenome alignments that can incorporate newly-sequenced assemblies. We previously developed Mumemto, which computes maximal unique matches (multi-MUMs) across pangenomes using compressed indexing. In this work, we extend Mumemto by introducing two new partitioning and merging strategies. Both strategies enable highly parallel, memory efficient, and updateable computation of multi-MUMs. One of the strategies, called string-based merging, is also capable of conducting the merges in a way that follows the shape of a phylogenetic tree, naturally yielding the multi-MUM for the tree's internal nodes as well as the root. With these strategies, Mumemto now scales to 474 human haplotypes, the only multi-MUM method able to do so. It also introduces a time-memory tradeoff that allows Mumemto to be tailored to more scenarios, including in resource-limited settings.
Figures

References
-
- Abouelhoda MI, Kurtz S Ohlebusch E 2004. “Replacing suffix trees with enhanced suffix arrays”. Journal of Discrete Algorithms 2: pp. 53–86
-
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS Haussler D 2004. “Ultraconserved elements in the human genome”. Science 304: pp. 1321–1325 - PubMed
-
- Brown NK, Shivakumar VS Langmead B (2025), “Improved Pangenomic Classification Accuracy with Chain Statistics”, in: Research in Computational Molecular Biology, ed. by Sankararaman S, Cham: Springer Nature Switzerland, pp. 190–208
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources