

This is a preprint.
Efficient profiling of total RNA in single cells with STORM-seq
- PMID: 40475517
- PMCID: PMC12139823
- DOI: 10.1101/2022.03.14.484332
Efficient profiling of total RNA in single cells with STORM-seq
Abstract
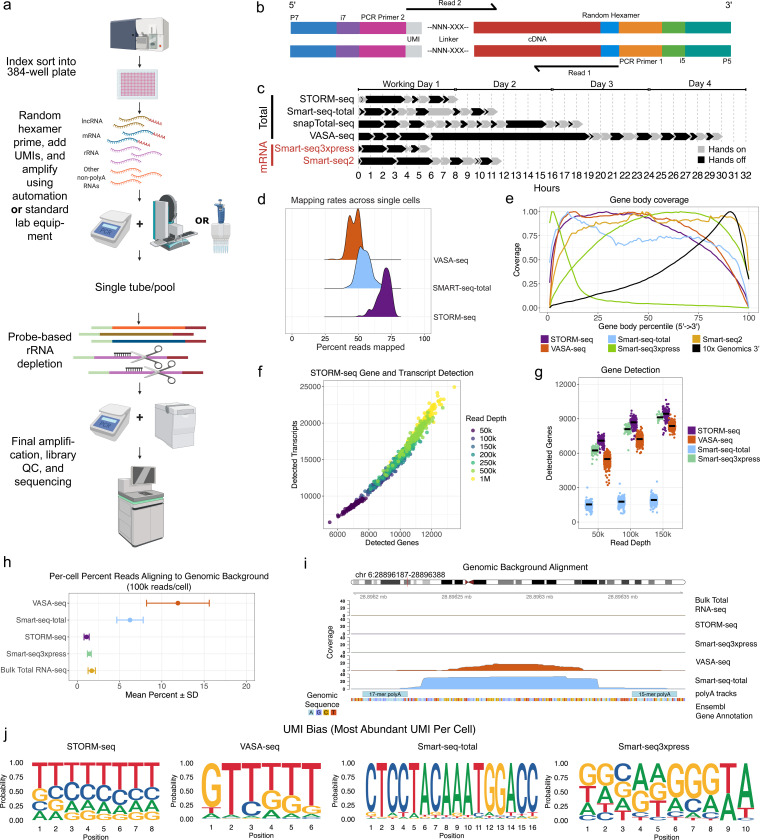
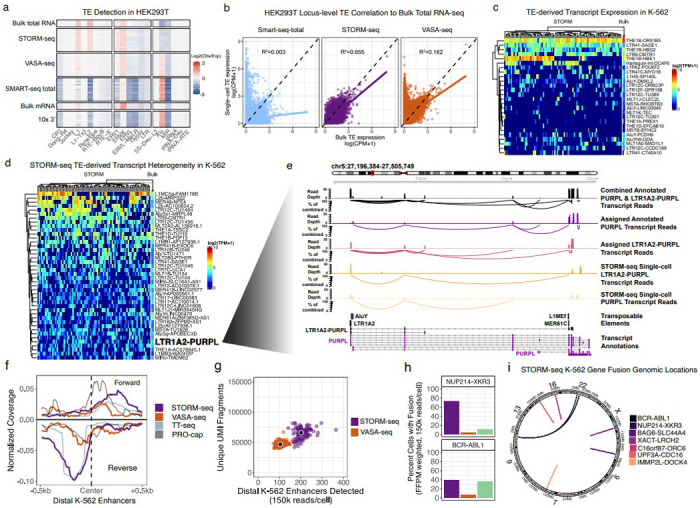
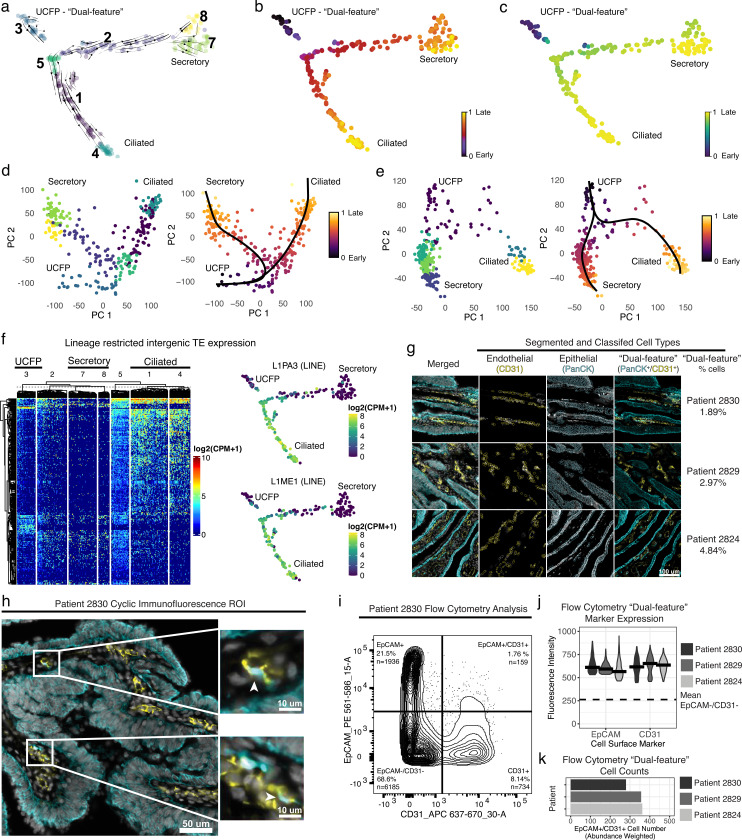
Despite significant advances, current single-cell RNA sequencing (scRNA-seq) technologies often struggle with accurately detecting non-coding transcripts, achieving full-length RNA coverage, and/or resolving transcript-level complexity. Many are also difficult to implement or inaccessible without specialized liquid handlers, further limiting their utility. We present Single-cell TOtal RNA-seq Miniaturized (STORM-seq), a random-hexamer primed, ribo-reduced single-cell total RNA sequencing (sc-total-RNA-seq) protocol using standard laboratory equipment. Adapted as a kit, STORM-seq constructs sequence-ready libraries in one working day, producing the highest complexity scRNA-seq libraries to-date, robustly measuring transcript isoforms and clinically relevant gene fusions in single cells. STORM-seq faithfully reconstructs expression profiles of locus-level transposable elements (TEs), and provides high-resolution profiling of transient, low-abundance enhancer RNAs (eRNAs), offering a powerful tool to dissect single-cell gene regulatory networks in unprecedented detail. Applied to human fallopian tube epithelium, the improved transcriptional resolution reveals a putative progenitor-like population and intermediate cell states, shaped by TEs and non-coding RNAs.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources