

This is a preprint.
Decoding protein-peptide interactions using a large, target-agnostic yeast surface display library
- PMID: 40475574
- PMCID: PMC12139779
- DOI: 10.1101/2025.05.19.654863
Decoding protein-peptide interactions using a large, target-agnostic yeast surface display library
Update in
-
Decoding Protein-Peptide Interactions Using a Large, Target-Agnostic Yeast Surface Display Library.ACS Chem Biol. 2025 Aug 13. doi: 10.1021/acschembio.5c00265. Online ahead of print. ACS Chem Biol. 2025. PMID: 40804242
Abstract
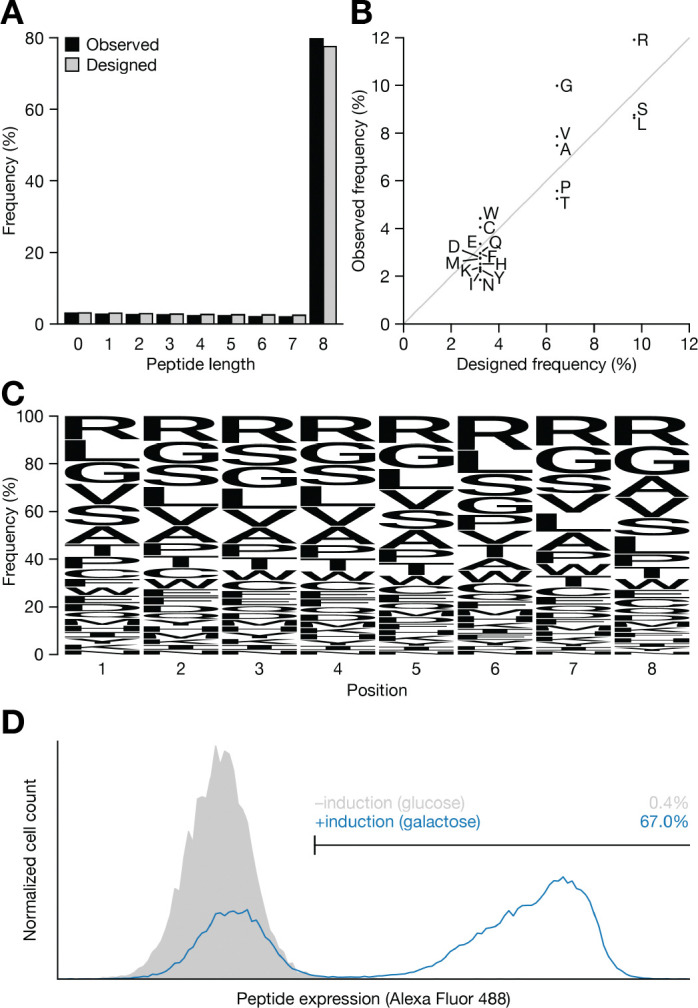
Protein-peptide interactions underlie key biological processes and are commonly utilized in biomedical research and therapeutic discovery. It is often desirable to identify peptide sequence properties that confer high-affinity binding to a target protein. However, common approaches to such characterization are typically low throughput and only sample regions of sequence space near an initial hit. To overcome these challenges, we built a yeast surface displayed library representing ~6.1 × 109 unique peptides. We then performed screens against diverse protein targets, including two antibodies, an E3 ubiquitin ligase, and an essential membrane-bound bacterial enzyme. In each case, we observed motifs that appear to drive peptide binding and we identified multiple novel, high-affinity clones. These results highlight the library's utility as a robust and versatile tool for discovering peptide ligands and for characterizing protein-peptide binding interactions more generally. To enable further studies, we will make the library freely available upon request.
Conflict of interest statement
Competing interests A.C.K. is a cofounder and consultant for Tectonic Therapeutic and Seismic Therapeutic, and for the Institute for Protein Innovation, a nonprofit research institute. The remaining authors declare no competing interests.
Figures





Similar articles
-
Decoding Protein-Peptide Interactions Using a Large, Target-Agnostic Yeast Surface Display Library.ACS Chem Biol. 2025 Aug 13. doi: 10.1021/acschembio.5c00265. Online ahead of print. ACS Chem Biol. 2025. PMID: 40804242
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
-
Aspects of Genetic Diversity, Host Specificity and Public Health Significance of Single-Celled Intestinal Parasites Commonly Observed in Humans and Mostly Referred to as 'Non-Pathogenic'.APMIS. 2025 Sep;133(9):e70036. doi: 10.1111/apm.70036. APMIS. 2025. PMID: 40923351 Free PMC article. Review.
-
Identification of novel, clinically correlated autoantigens in the monogenic autoimmune syndrome APS1 by proteome-wide PhIP-Seq.Elife. 2020 May 15;9:e55053. doi: 10.7554/eLife.55053. Elife. 2020. PMID: 32410729 Free PMC article.
-
The quantity, quality and findings of network meta-analyses evaluating the effectiveness of GLP-1 RAs for weight loss: a scoping review.Health Technol Assess. 2025 Jun 25:1-73. doi: 10.3310/SKHT8119. Online ahead of print. Health Technol Assess. 2025. PMID: 40580049 Free PMC article.
References
-
- London N., Raveh B. & Schueler-Furman O. Druggable protein-protein interactions--from hot spots to hot segments. Curr. Opin. Chem. Biol. 17, 952–959 (2013). - PubMed
-
- Houen G. Peptide Antibodies: Past, Present, and Future: Methods and Protocols. in Peptide Antibodies (ed. Houen G.) vol. 1348 1–6 (Springer New York, New York, NY, 2015). - PubMed
-
- Berg K. M. C. W. Janeway’s Immunobiology, 10th Edition. (W. W. Norton & Company, NewYork, NY, 2022).
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources