

This is a preprint.
A Tandem Repeat Atlas for the Genome of Inbred Mouse Strains: A Genetic Variation Resource
- PMID: 40475611
- PMCID: PMC12139781
- DOI: 10.1101/2025.05.23.655792
A Tandem Repeat Atlas for the Genome of Inbred Mouse Strains: A Genetic Variation Resource
Abstract
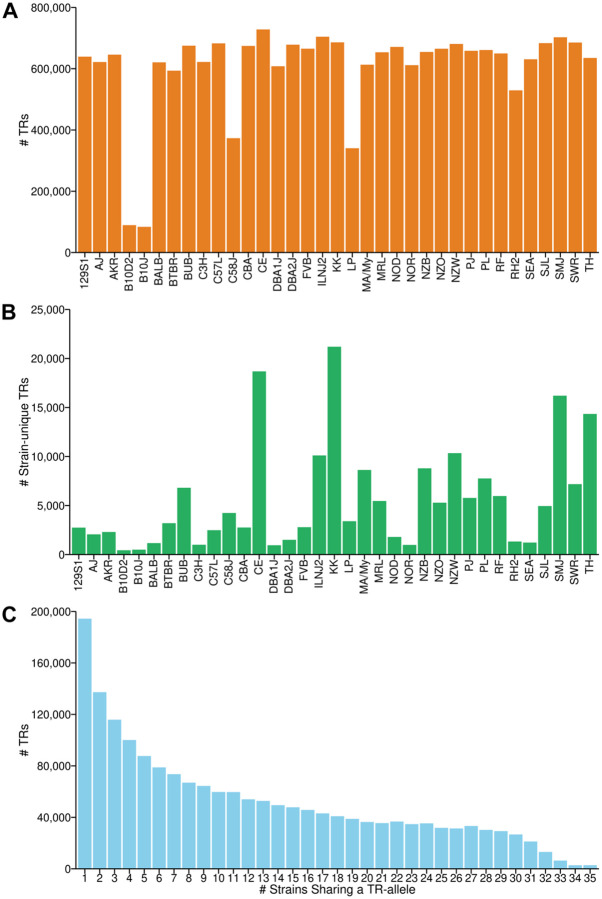
Tandem repeats (TRs) are a significant source of genetic variation in the human population; and TR alleles are responsible for over 60 human genetic diseases and for inter-individual differences in many biomedical traits. Therefore, we utilized long-read sequencing and state of the art computational programs to produce a database with 2,528,854 TRs covering 39 inbred mouse strains. As in humans, murine TRs are abundant and were primarily located in intergenic regions. However, there were important species differences: murine TRs did not have the extensive number of repeat expansions like those associated with human repeat expansion diseases and they were not associated with transposable elements. We demonstrate by analysis of two biomedical phenotypes, which were identified over 40 years ago, that this TR database can enhance our ability to characterize the genetic basis for trait differences among the inbred strains.
Conflict of interest statement
DECLARATION OF INTERESTS W.R., W.L., Z.F, B.W., Z.C., and G.P. declare no conflict of interest. E.D. is an employee and shareholder of Pacific Biosciences.
Figures




Similar articles
-
Large scale in silico characterization of repeat expansion variation in human genomes.Sci Data. 2020 Sep 8;7(1):294. doi: 10.1038/s41597-020-00633-9. Sci Data. 2020. PMID: 32901039 Free PMC article.
-
Large tandem repeats of grass frog (Rana temporaria) in silico and in situ.BMC Genomics. 2025 May 6;26(1):445. doi: 10.1186/s12864-025-11643-5. BMC Genomics. 2025. PMID: 40329174 Free PMC article.
-
Advancing genomic technologies and clinical awareness accelerates discovery of disease-associated tandem repeat sequences.Genome Res. 2022 Jan;32(1):1-27. doi: 10.1101/gr.269530.120. Epub 2021 Dec 29. Genome Res. 2022. PMID: 34965938 Free PMC article. Review.
-
A genome-wide spectrum of tandem repeat expansions in 338,963 humans.Cell. 2024 Apr 25;187(9):2336-2341.e5. doi: 10.1016/j.cell.2024.03.004. Epub 2024 Apr 5. Cell. 2024. PMID: 38582080 Free PMC article.
-
Statistical approaches to detecting and analyzing tandem repeats in genomic sequences.Front Bioeng Biotechnol. 2015 Mar 17;3:31. doi: 10.3389/fbioe.2015.00031. eCollection 2015. Front Bioeng Biotechnol. 2015. PMID: 25853125 Free PMC article. Review.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials