

Origin of Neisseria meningitidis clonal complex 4821
- PMID: 40476506
- PMCID: PMC12180329
- DOI: 10.1080/22221751.2025.2515461
Origin of Neisseria meningitidis clonal complex 4821
Abstract
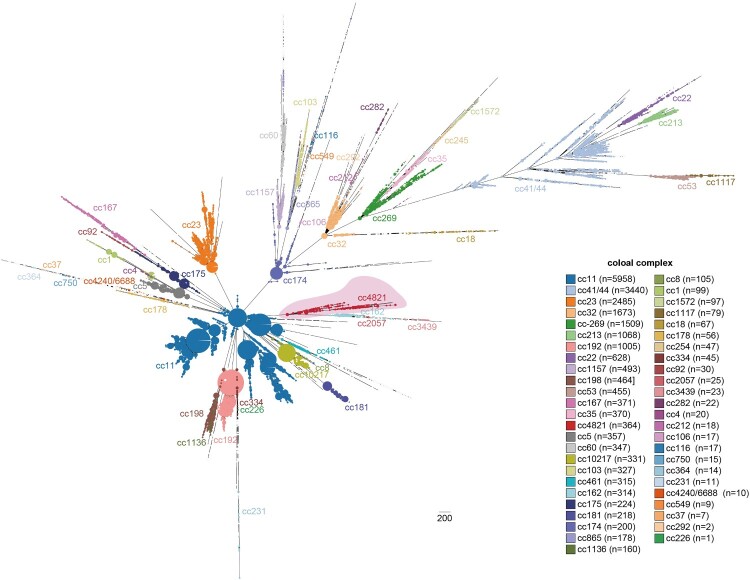
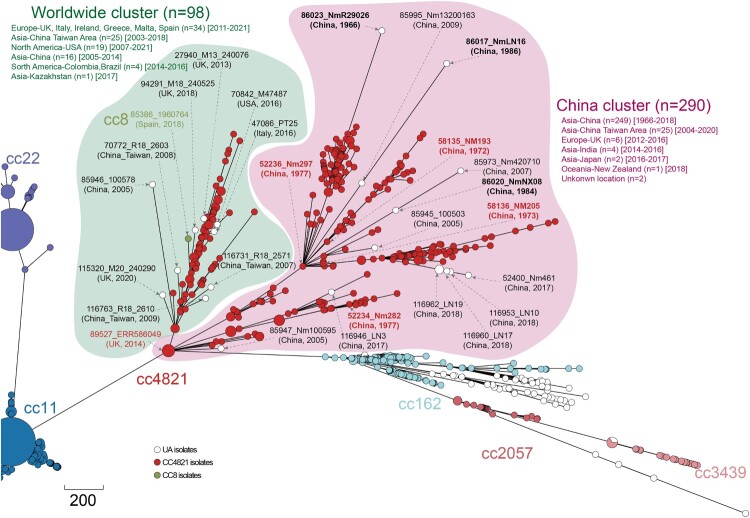
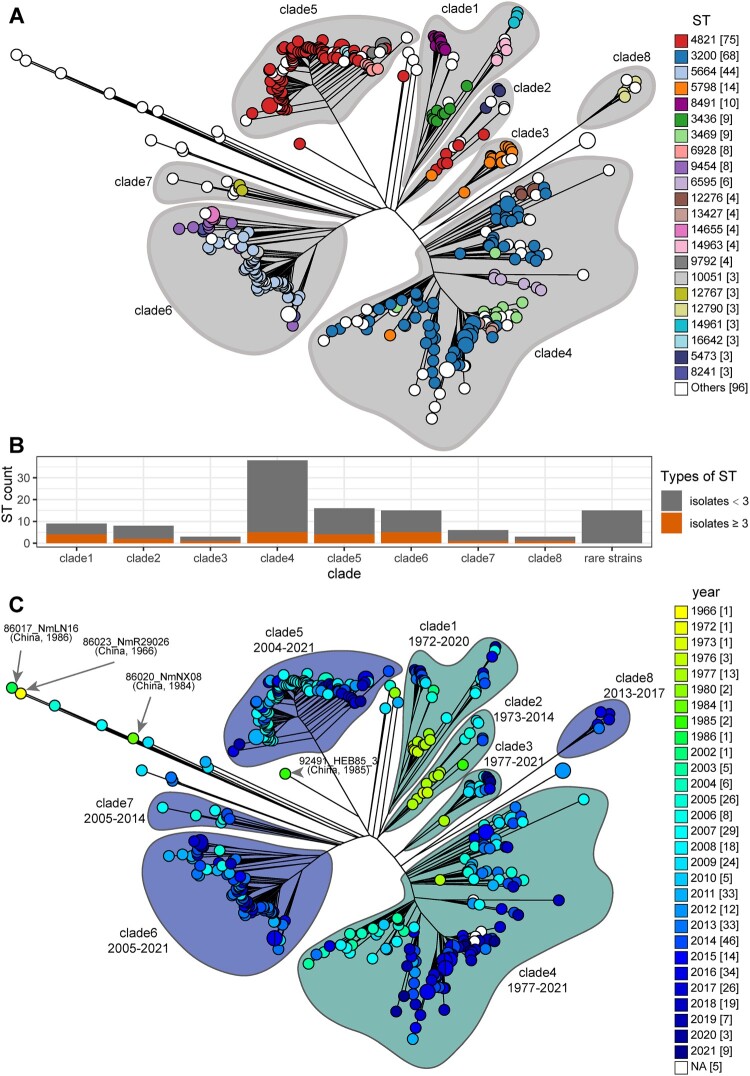
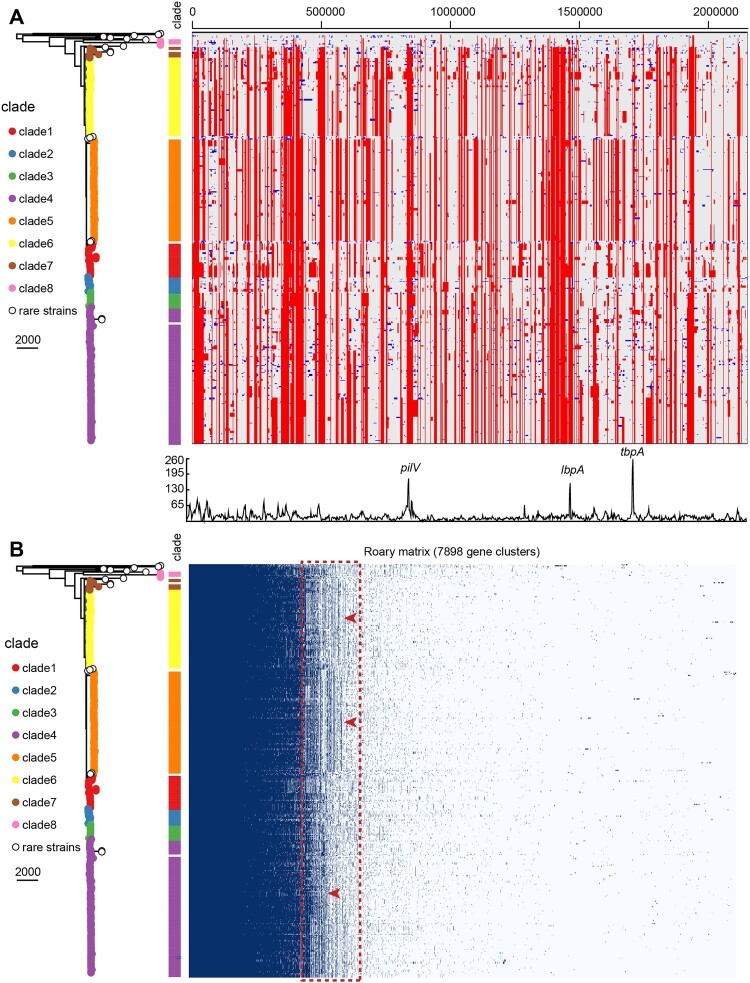
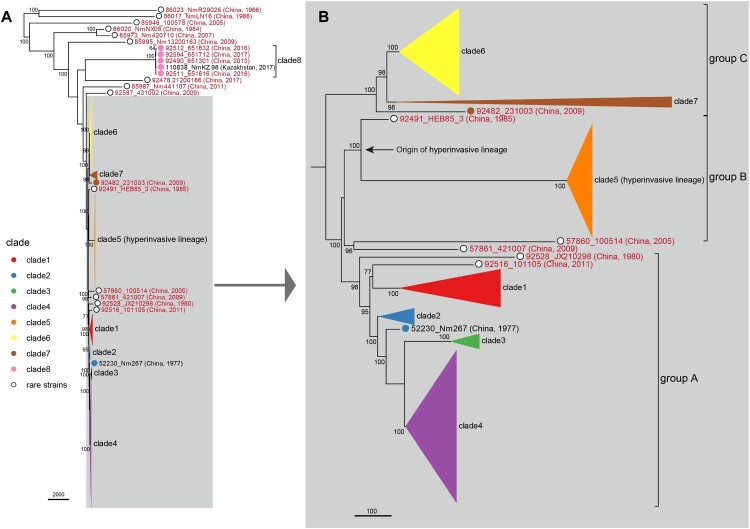
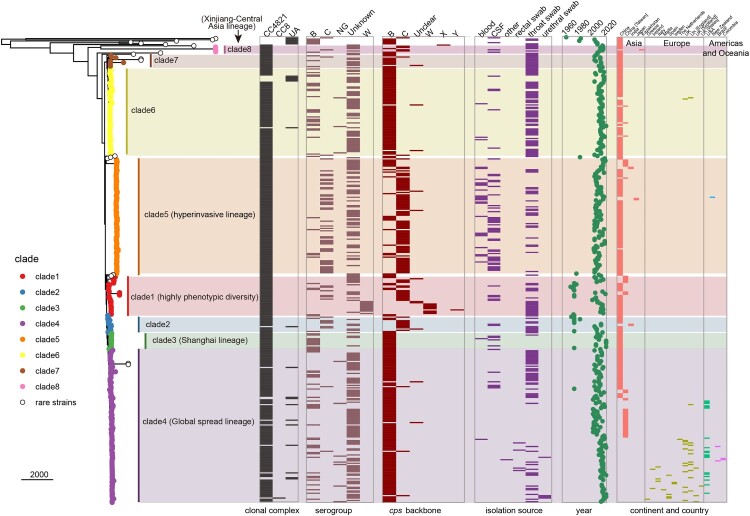
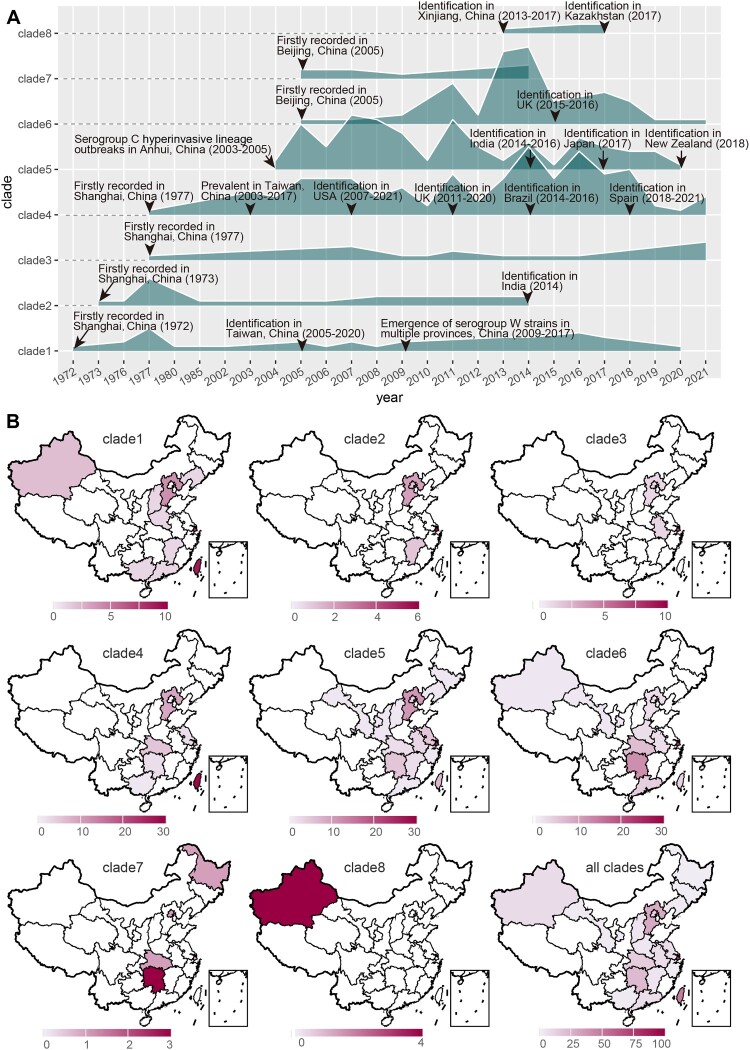
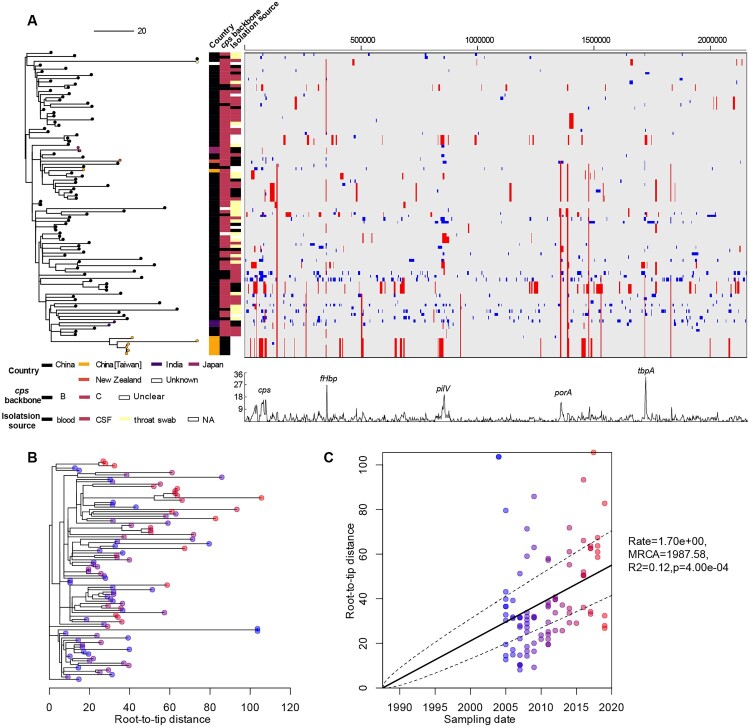
Neisseria meningitidis (N. meningitidis) is the causative agent of human invasive meningococcal disease (IMD). Clonal complex (CC) 4821 is a unique genetic cluster of N. meningitidis that emerged two decades ago in Anhui Province, China and became the predominant cluster. However, the evolutionary origin of CC4821 remains unclear. Herein, a distinct CC4821 clade was identified by a comprehensive cgMLST analysis of 26,801 N. meningitidis genomes. The CC4821 clade comprised 388 N. meningitidis isolates, with 364 assigned to CC4821, 1 assigned to CC8, and 23 unassigned (UA), as they could not be assigned to any defined CC. The phylogenetic analysis of the CC4821 clade revealed that six UA isolates, including the UA isolate NmR29026 collected in 1966 from Liaoning Province, China, occupied a basal position compared to all isolates within the CC4821 clade, indicating that CC4821 originated in the 1960s. Eight subclades (clades 1-8) were recognized within the CC4821 clade. Clades 1-4 have been present since the 1970s, while clades 5-8 emerged after the 2000s. Clade 5 represents a hyperinvasive lineage. N. meningitidis isolate HEB85-3, collected in 1985 in Hebei Province, China, exhibited the closest evolutionary relationship to clade 5, suggesting it is related to the origin of this hyperinvasive lineage. Our study reveals that CC4821 has emerged as the predominant cluster of N. meningitidis in China, representing the culmination of at least 60 years of continuous evolution in China, and is not solely attributable to the outbreak two decades ago.
Keywords: Neisseria meningitidis; bacterial evolution; clonal complex 4821; hyperinvasive lineage; population structure.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures
References
-
- Andam CP, Challagundla L, Azarian T, et al. Population structure of pathogenic bacteria. In: Tibayrenc M, editor. Genetics and evolution of infectious diseases. 2nd ed. London: Elsevier; 2017. p. 51–70.
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical