

Gasdermin E deficiency limits inflammation and lung damage during influenza virus infection
- PMID: 40481027
- PMCID: PMC12144252
- DOI: 10.1038/s41419-025-07748-0
Gasdermin E deficiency limits inflammation and lung damage during influenza virus infection
Abstract
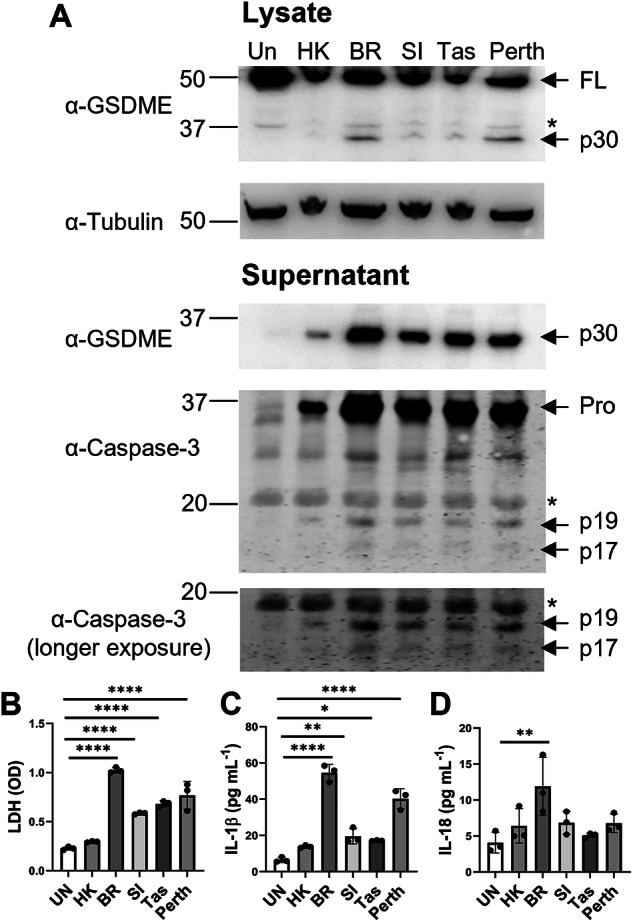
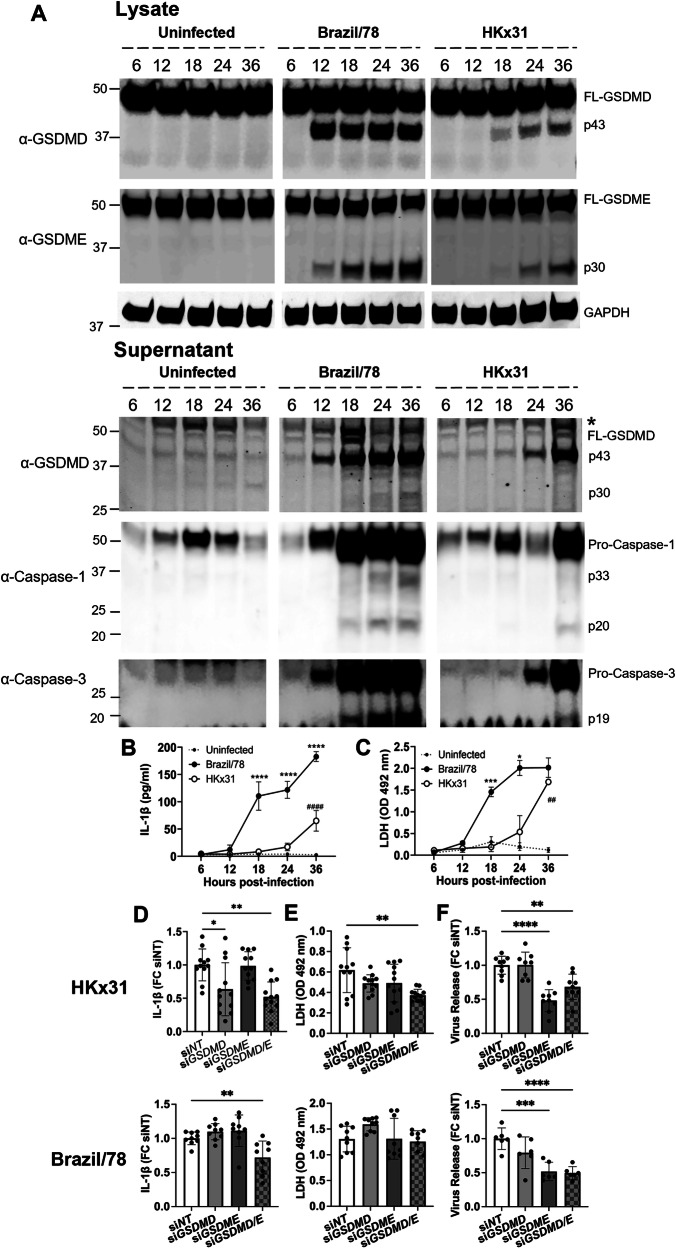
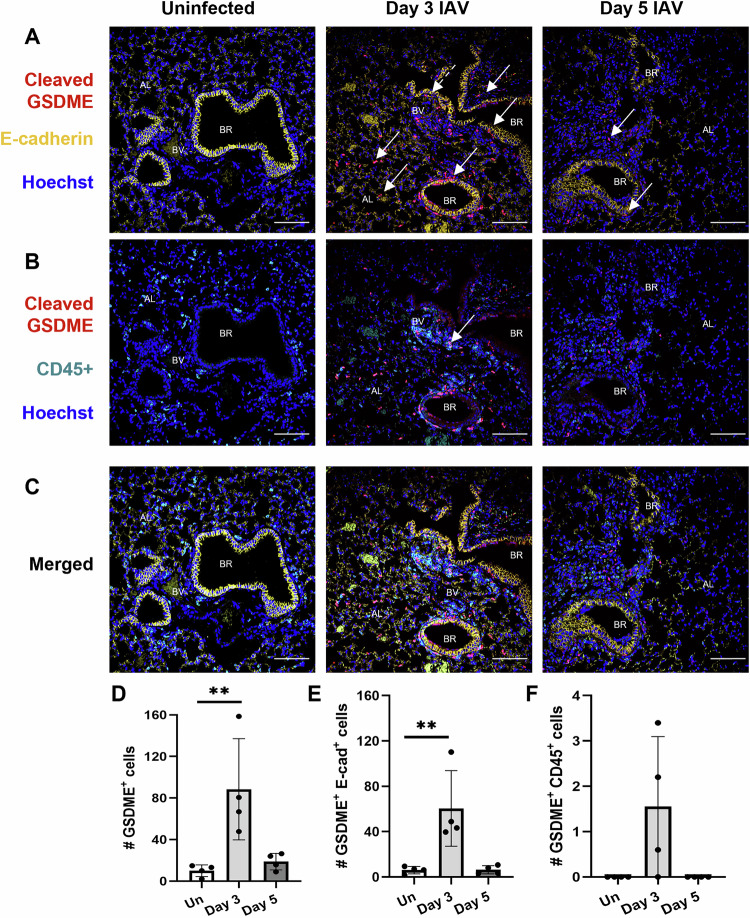
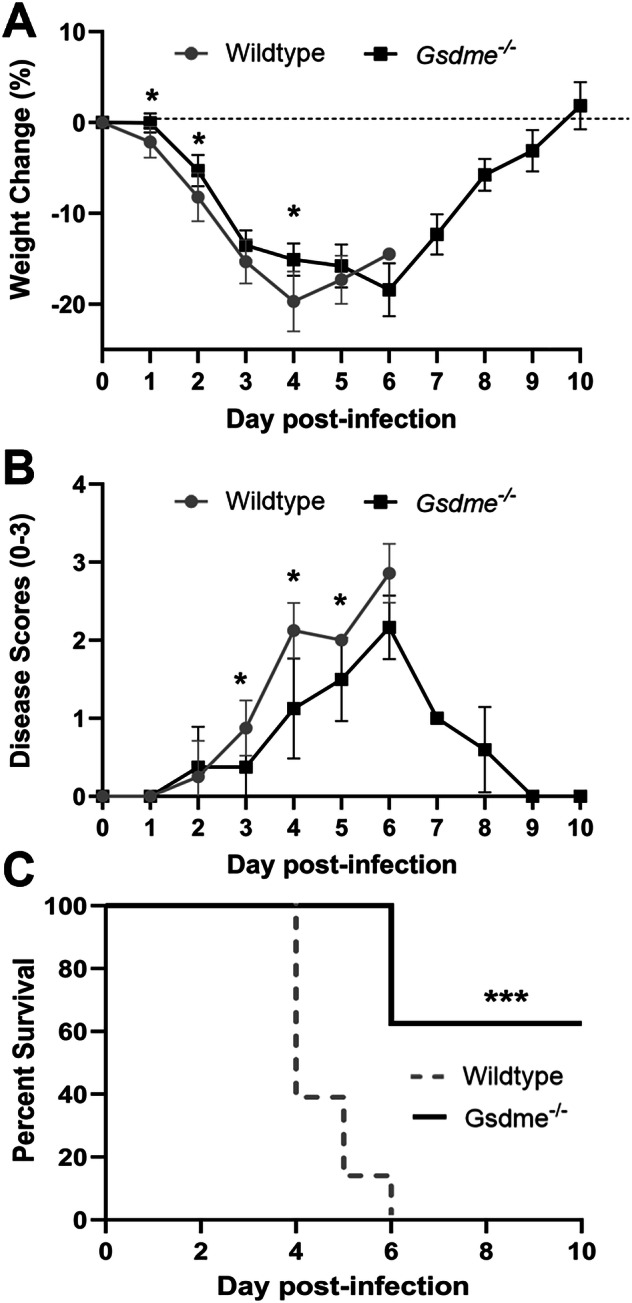
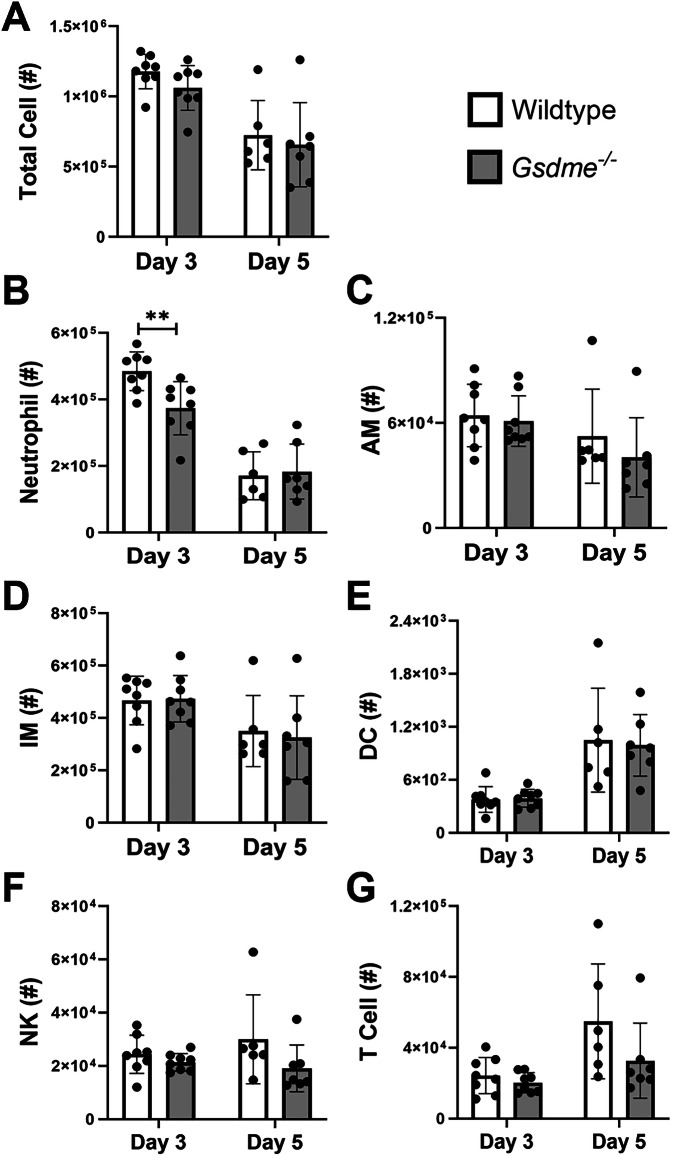
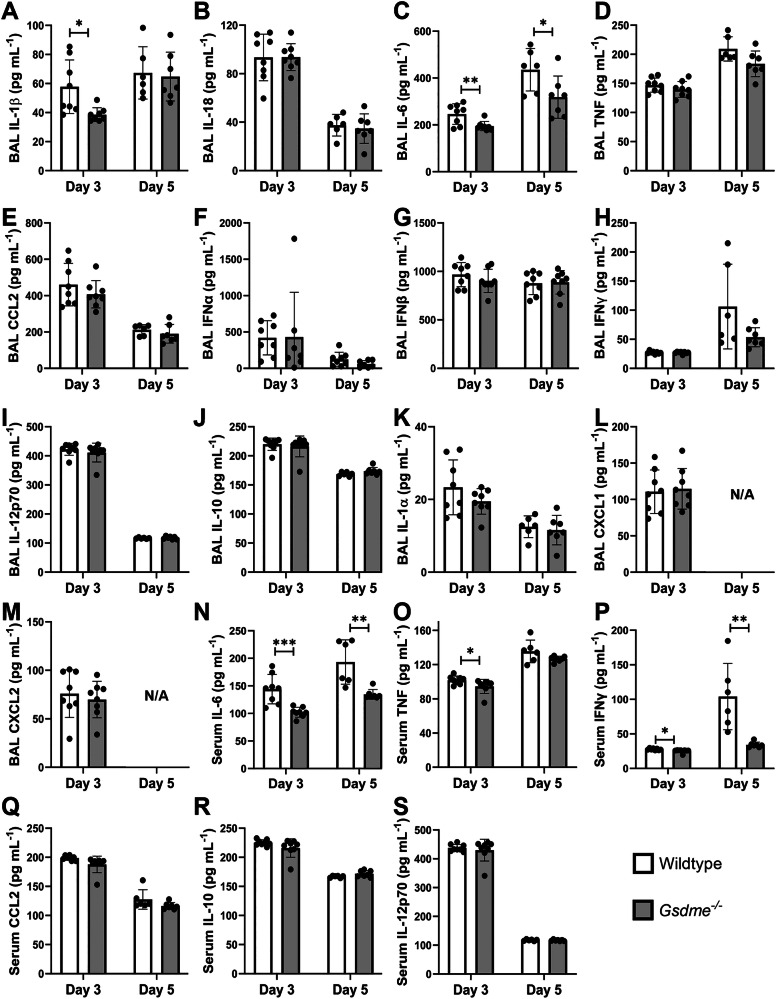
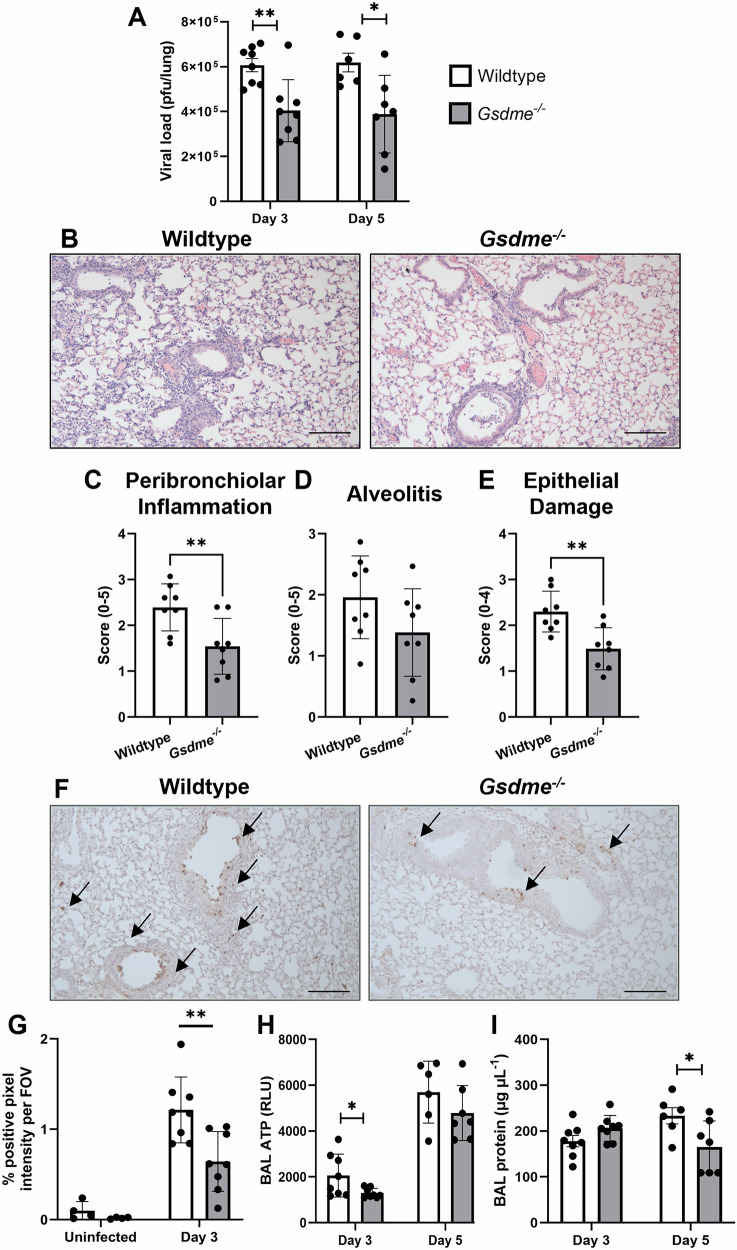
Severe influenza A virus (IAV) infections are associated with hyperinflammation and significant lung damage. Gasdermin E (GSDME) mediates pyroptosis, a lytic and inflammatory type of cell death. Cleavage of GSDME by caspase-3 releases the active N-terminal domain, which subsequently forms transmembrane pores, leading to cell lysis and death. In this study, we investigated a role for GSDME in severe influenza. Infection of human bronchial epithelial cells revealed that IAV induces GSDME cleavage and activation, with the magnitude and kinetics of GSDME activation differing between IAV strains. Caspase-3-mediated GSDME activation preceded and overwhelmed gasdermin D (GSDMD) activation. siRNA silencing in vitro confirmed both gasdermins are active in human bronchial epithelial cells and cooperate to drive IAV responses. IAV infection of mice promoted GSDME cleavage in E-cadherin+ epithelial cells in vivo at day 3. Mice deficient in GSDME (Gsdme-/-) showed improved survival and greater influenza disease resistance compared to their wildtype littermate controls. Gsdme-/- mice exhibited reduced neutrophil infiltration and levels of cytokines IL-6 and IL-1β in the airways and IL-6, TNF, and IFNγ in the serum. This was accompanied by reduced viral loads, lung pathology, and epithelial cell death. Together, these findings demonstrate a pivotal role for GSDME in severe influenza pathogenesis.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Gasdermin D promotes hyperinflammation and immunopathology during severe influenza A virus infection.Cell Death Dis. 2023 Nov 9;14(11):727. doi: 10.1038/s41419-023-06258-1. Cell Death Dis. 2023. PMID: 37945599 Free PMC article.
-
Gasdermin E-mediated keratinocyte pyroptosis participates in the pathogenesis of psoriasis by promoting skin inflammation.Br J Dermatol. 2024 Aug 14;191(3):385-396. doi: 10.1093/bjd/ljae179. Br J Dermatol. 2024. PMID: 38655652
-
Caspase 3/GSDME-Mediated Corneal Epithelial Pyroptosis Promotes Dry Eye Disease.Invest Ophthalmol Vis Sci. 2025 Jan 2;66(1):24. doi: 10.1167/iovs.66.1.24. Invest Ophthalmol Vis Sci. 2025. PMID: 39792075 Free PMC article.
-
The pyroptotic role of Caspase-3/GSDME signalling pathway among various cancer: A Review.Int J Biol Macromol. 2023 Jul 1;242(Pt 2):124832. doi: 10.1016/j.ijbiomac.2023.124832. Epub 2023 May 15. Int J Biol Macromol. 2023. PMID: 37196719 Review.
-
Indoleamine 2,3-dioxygenase 1 drives epithelial cells ferroptosis in influenza-induced acute lung injury.Redox Biol. 2025 Apr;81:103572. doi: 10.1016/j.redox.2025.103572. Epub 2025 Feb 26. Redox Biol. 2025. PMID: 40023977 Free PMC article. Review.
Cited by
-
Gasdermin E is Dispensable for H1N1 Influenza Virus Pathogenesis in Mice.bioRxiv [Preprint]. 2025 Jul 29:2025.07.29.667514. doi: 10.1101/2025.07.29.667514. bioRxiv. 2025. PMID: 40766393 Free PMC article. Preprint.
References
-
- Nguyen THO, Rowntree LC, Chua BY, Thwaites RS, Kedzierska K. Defining the balance between optimal immunity and immunopathology in influenza virus infection. Nat Rev Immunol. 2024;24:720–35. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
