

COL4A5-p.Gly624Asp is the Predominant Variant in Europe Associated With a Mild Alport Syndrome Phenotype
- PMID: 40485705
- PMCID: PMC12142618
- DOI: 10.1016/j.ekir.2025.02.031
COL4A5-p.Gly624Asp is the Predominant Variant in Europe Associated With a Mild Alport Syndrome Phenotype
Abstract
Introduction: Pathogenic variants in COL4A3-5 are common causes of inherited kidney disease. The clinical presentation extends from classical Alport syndrome (AS) to focal segmental glomerulosclerosis (FSGS) without extrarenal manifestation. In this study, we aimed to assess the genetic and phenotypic spectrum, along with the associated natural histories, in a cohort of patients with AS from 3 tertiary centers in Central Europe.
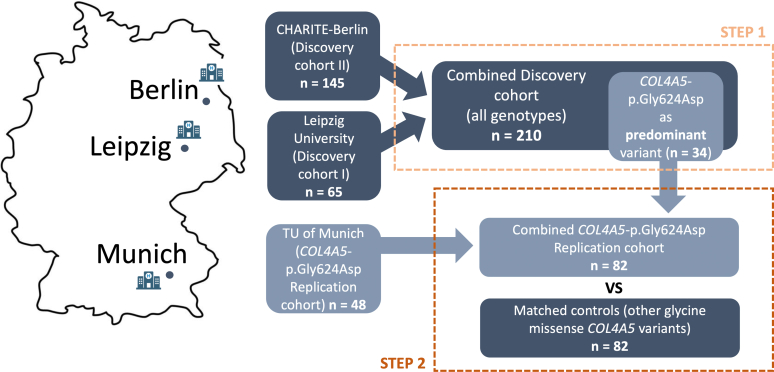
Methods: A total of 210 patients with disease causing variants in one of the COL4A3-5 genes were characterized and evaluated for genotype-phenotype correlations. In addition, 48 COL4A5-p.Gly624Asp carriers were analyzed for replication and pooled analysis.
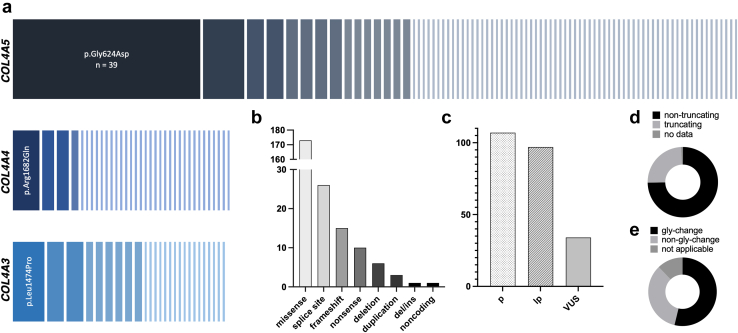
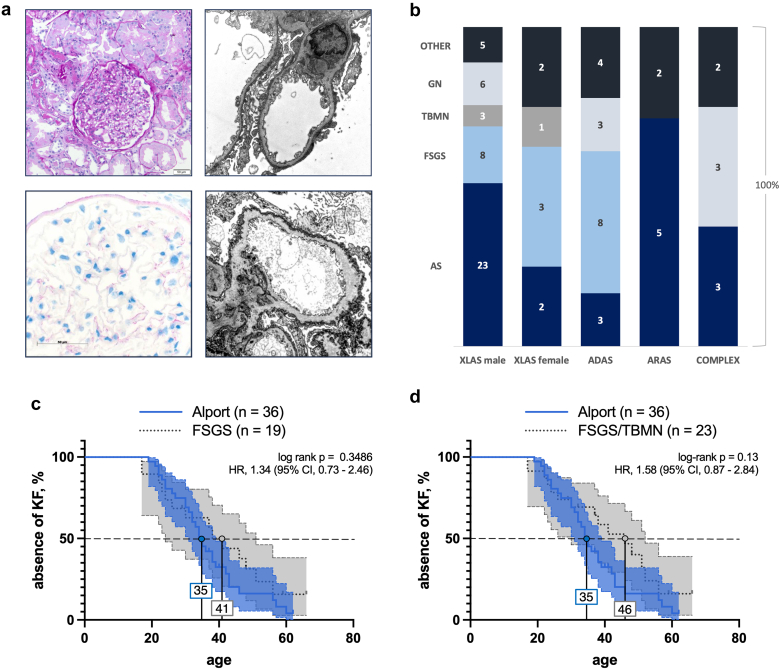
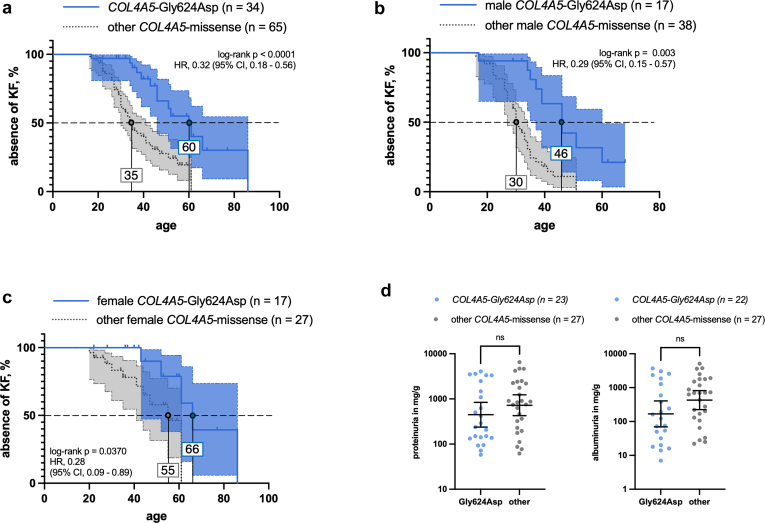
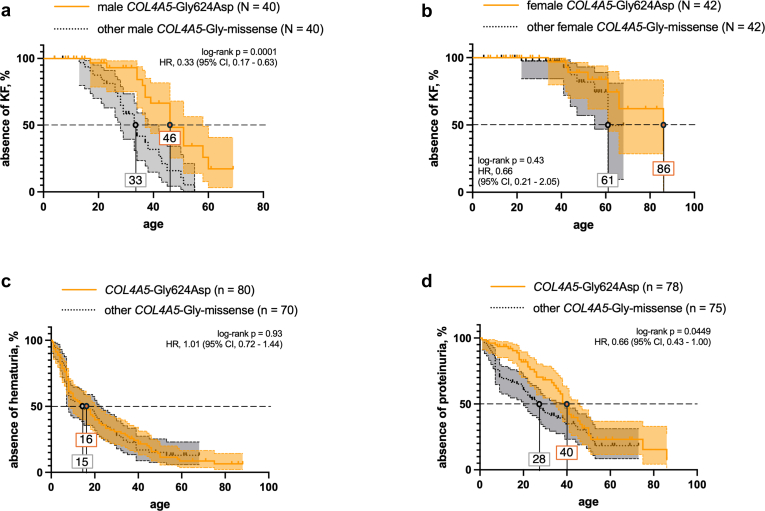
Results: COL4A5-p.Gly624Asp was by far the most common variant, accounting for 16% of all genetic diagnoses. These patients presented with overall milder renal phenotypes than patients with other COL4A5 missense variants and COL4A5 glycine-missense variants after age- and sex-matching. In patients lacking a wild-type allele (X-Linked AS [XLAS] males and autosomal recessive AS [ARAS]), histological AS was most frequently observed in kidney biopsies, and truncating variants were associated with increased disease severity. Conversely, in patients with a wild-type allele present (XLAS females and autosomal dominant AS [ADAS]), FSGS was more frequently observed. Predicted protein truncation was not inferior to missense alterations in terms of renal survival.
Conclusion: The predominance of the European COL4A5 founder variant p.Gly624Asp allowed for the creation of the largest cohort of patients with an identical Alport variant to date, confirming the more favorable renal prognosis specific to this amino acid change. Allelic and gene dosage effects drive phenotypic differences and should be incorporated into future risk models.
Keywords: COL4A3; COL4A4; COL4A5; alport syndrome; chronic kidney disease; collagen type IV.
© 2025 International Society of Nephrology. Published by Elsevier Inc.
Figures


References
-
- Mariyama M., Leinonen A., Mochizuki T., Tryggvason K., Reeders S.T. Complete primary structure of the human alpha3(IV) collagen chain. Coexpression of the alpha 3(IV) and alpha 4(IV) collagen chains in human tissues. J Biol Chem. 1994;269:23013–23017. doi: 10.1016/S0021-9258(17)31612-5. - DOI - PubMed
-
- Hostikka S.L., Eddy R.L., Byers M.G., Höyhtyä M., Shows T.B., Tryggvason K. Identification of a distinct type IV collagen alpha chain with restricted kidney distribution and assignment of its gene to the locus of X chromosome-linkedAlport syndrome. Proc Natl Acad Sci U S A. 1990;87:1606–1610. doi: 10.1073/pnas.87.4.1606. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
