

Adiponectin-receptor agonism prevents right ventricular tissue pathology in a mouse model of Duchenne muscular dystrophy
- PMID: 40490137
- PMCID: PMC12274718
- DOI: 10.1016/j.molmet.2025.102179
Adiponectin-receptor agonism prevents right ventricular tissue pathology in a mouse model of Duchenne muscular dystrophy
Abstract
Objective: Cardiac fibrosis during Duchenne muscular dystrophy (DMD) arises from cellular damage and inflammation and is associated with metabolic dysfunction. The extent to which these relationships develop across all 4 cardiac chambers, particularly during early-stage disease, remains unknown.
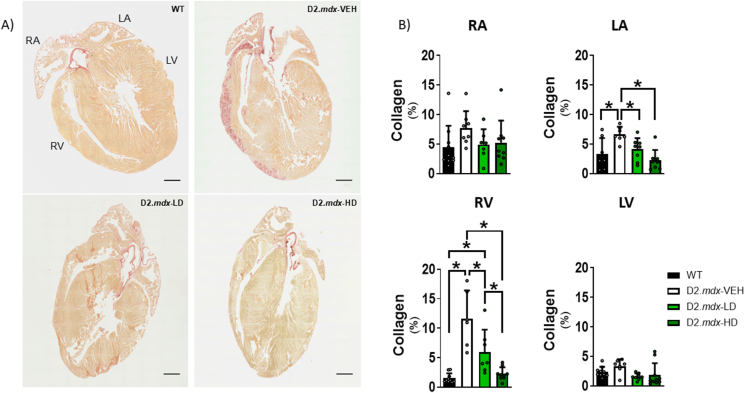
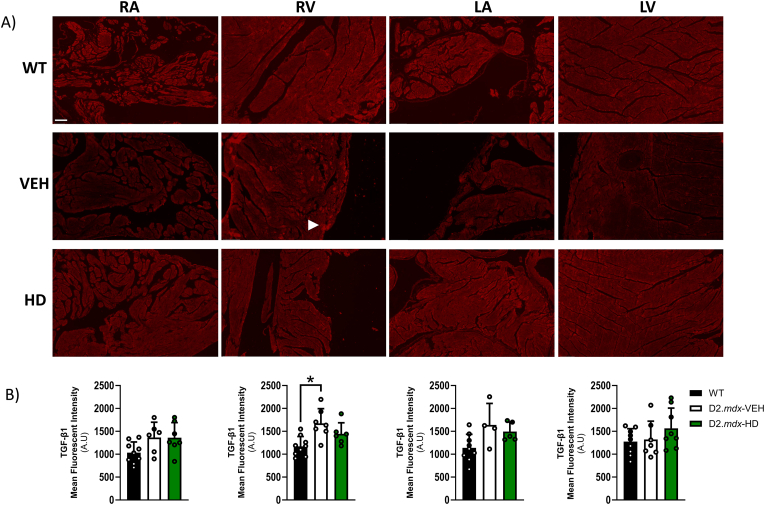
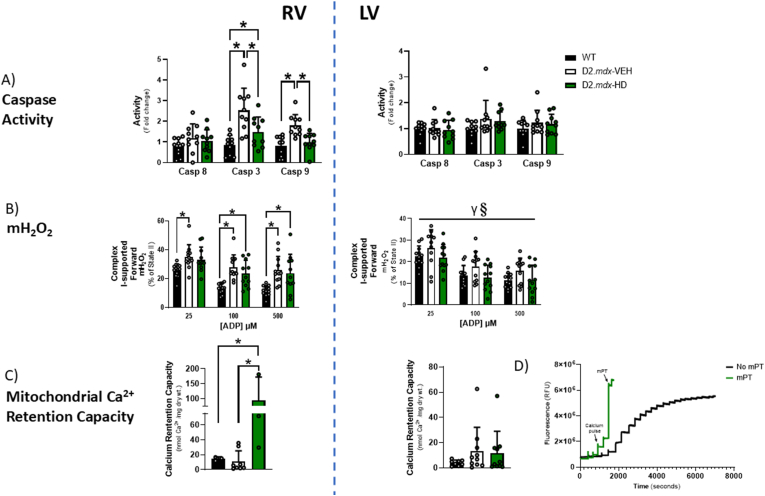
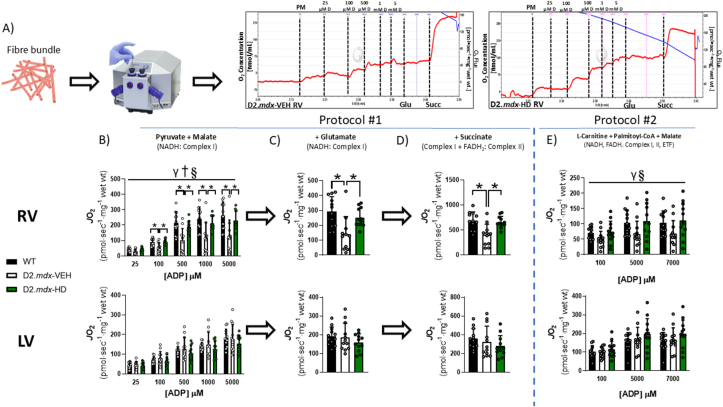
Methods and results: We discovered that very young D2.mdx mice exhibit fibrosis exclusively in the right ventricle (RV) and left atrium. Concurrent myocardial disorganization in the RV was related to a highly specific inflammatory signature of increased infiltrating pro-inflammatory macrophages (CD11b+CD45+CD64+F4/80+CCR2+), myofibre mitochondrial-linked apoptosis, and reduced carbohydrate and fat oxidation. This relationship did not occur in the left ventricle. Short-term daily administration of a peptidomimetic adiponectin receptor agonist, ALY688, prevented RV fibrosis, infiltrating macrophages, and mitochondrial stress as well as left atrial fibrosis.
Conclusions: Our discoveries demonstrate early-stage cardiac tissue pathology occurs in a chamber-specific manner and is prevented by adiponectin receptor agonism, thereby opening a new direction for developing therapies that prevent tissue remodeling during DMD.
Keywords: Cardiomyopathy; Duchenne muscular dystrophy; Fibrosis; Inflammation; Mitochondria.
Copyright © 2025 The Author(s). Published by Elsevier GmbH.. All rights reserved.
Conflict of interest statement
Declaration of competing interest Authors declare that they have no competing interests. HHH is an employee of Allysta and GS and AAS consult for Allysta.
Figures














References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
