

Chromothripsis-associated chromosome 21 amplification orchestrates transformation to blast-phase MPN through targetable overexpression of DYRK1A
- PMID: 40490510
- PMCID: PMC12165854
- DOI: 10.1038/s41588-025-02190-6
Chromothripsis-associated chromosome 21 amplification orchestrates transformation to blast-phase MPN through targetable overexpression of DYRK1A
Abstract
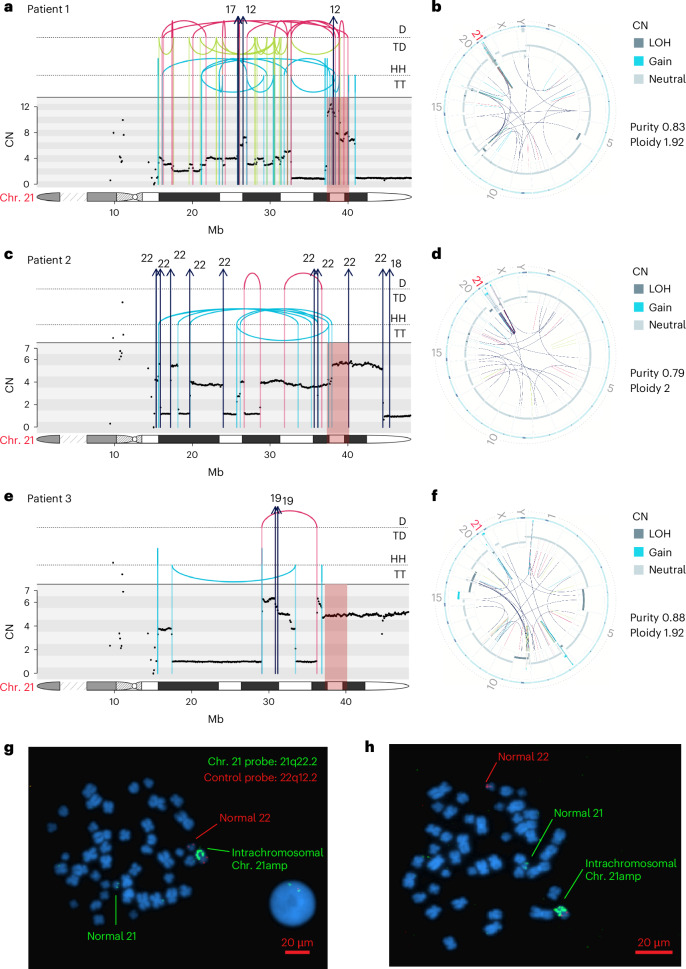
Chromothripsis, the chaotic shattering and repair of chromosomes, is common in cancer. Whether chromothripsis generates actionable therapeutic targets remains an open question. In a cohort of 64 patients in blast phase of a myeloproliferative neoplasm (BP-MPN), we describe recurrent amplification of a region of chromosome 21q ('chr. 21amp') in 25%, driven by chromothripsis in a third of these cases. We report that chr. 21amp BP-MPN has a particularly aggressive and treatment-resistant phenotype. DYRK1A, a serine threonine kinase, is the only gene in the 2.7-megabase minimally amplified region that showed both increased expression and chromatin accessibility compared with non-chr. 21amp BP-MPN controls. DYRK1A is a central node at the nexus of multiple cellular functions critical for BP-MPN development and is essential for BP-MPN cell proliferation in vitro and in vivo, and represents a druggable axis. Collectively, these findings define chr. 21amp as a prognostic biomarker in BP-MPN, and link chromothripsis to a therapeutic target.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: A patent relating to the TARGET-seq technique is licensed to Alethiomics Ltd, a spin out company from the University of Oxford with equity owned by B.P. and A.J.M. E.P. is a founder, equity holder and holds a fiduciary role in Isabl Inc. A.J.C. is an employee of Owkin UK Ltd. The other authors declare no competing interests.
Figures














Update of
-
Chromothripsis orchestrates leukemic transformation in blast phase MPN through targetable amplification of DYRK1A.bioRxiv [Preprint]. 2023 Dec 10:2023.12.08.570880. doi: 10.1101/2023.12.08.570880. bioRxiv. 2023. Update in: Nat Genet. 2025 Jun;57(6):1478-1492. doi: 10.1038/s41588-025-02190-6. PMID: 38106192 Free PMC article. Updated. Preprint.
References
-
- Korbel, J. O. & Campbell, P. J. Criteria for inference of chromothripsis in cancer genomes. Cell10.1016/j.cell.2013.02.023 (2013). - PubMed
-
- Saha, C., Attwell, L., Harrison, C. N. & McLornan, D. P. Addressing the challenges of accelerated and blast phase myeloproliferative neoplasms in 2022 and beyond. Blood Rev.10.1016/j.blre.2022.100947 (2022). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
