

Alpelisib Therapy in 2 Patients With Congenital Hyperinsulinism
- PMID: 40492016
- PMCID: PMC12146258
- DOI: 10.1210/jcemcr/luaf099
Alpelisib Therapy in 2 Patients With Congenital Hyperinsulinism
Abstract
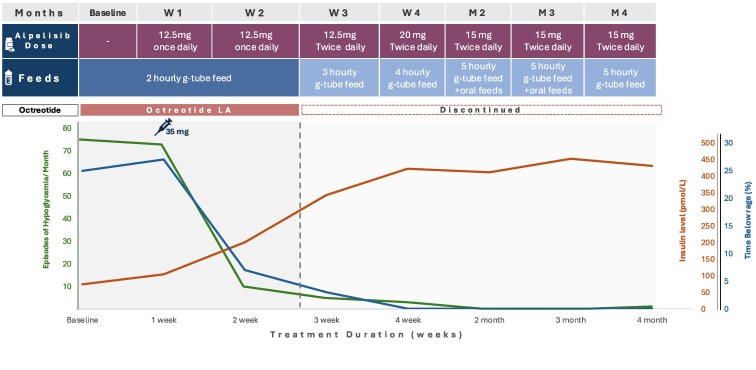
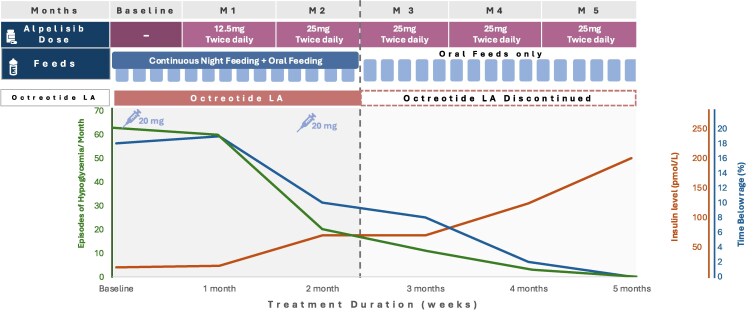
Congenital hyperinsulinism (CHI) is a disorder of unregulated insulin secretion, leading to severe hypoglycemia in most cases. We previously described the adjunct use of alpelisib therapy in a 3-month-old patient with CHI. We now describe our observations in 2 additional patients with severe CHI treated with alpelisib therapy, resulting in discontinuation of all existing treatments and normalization of feeding. Two children (aged 3 and 4 years) with CHI (homozygous ABCC8 and KCNJ11 pathological variants) who were unresponsive to conventional therapies were treated with alpelisib. Treatment was initiated at 12.5 mg daily, with gradual dose adjustments based on clinical responses. Outcome measures included blood glucose variability, frequency of hypoglycemic episodes, need for supplemental feeding, and treatment safety. In both cases, alpelisib significantly improved glucose levels, reducing the frequency of hypoglycemic episodes. This allowed for the tapering and discontinuation of other medications (diazoxide and octreotide) and facilitated a transition to bolus gastrostomy-tube/oral feeding. No significant adverse effects were reported. Alpelisib shows promise as both an adjunctive and primary therapy for CHI, improving glucose levels and reducing dependence on continuous feeding and other medications. Randomized controlled trials are needed to assess its long-term safety and efficacy for CHI.
Keywords: KCNJ11 pathological variant; Usher syndrome; alpelisib; congenital hyperinsulinism; hypoglycemia.
© The Author(s) 2025. Published by Oxford University Press on behalf of the Endocrine Society.
Figures
References
-
- Thornton PS, Stanley CA, De Leon DD. Congenital hyperinsulinism: an historical perspective. Horm Res Paediatr. 2022;95(6):631‐637. - PubMed
-
- Shah R, Harding J, Brown J, McKinlay C. Neonatal glycaemia and neurodevelopmental outcomes: a systematic review and meta-analysis. Neonatology. 2019;115(2):116‐126. - PubMed
-
- Hussain K. Diagnosis and management of hyperinsulinaemic hypoglycaemia of infancy. Horm Res Paediatr. 2008;69(1):2‐13. - PubMed
-
- Gillis D. Nonsyndromic genetic hyperinsulinism overview. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews® [Internet]. University of Washington; 2024.
Publication types
LinkOut - more resources
Full Text Sources