

The potential mechanisms of reciprocal regulation of gut microbiota-liver immune signaling in metabolic dysfunction-associated steatohepatitis revealed in multi-omics analysis
- PMID: 40492756
- PMCID: PMC12282060
- DOI: 10.1128/msystems.00518-25
The potential mechanisms of reciprocal regulation of gut microbiota-liver immune signaling in metabolic dysfunction-associated steatohepatitis revealed in multi-omics analysis
Abstract
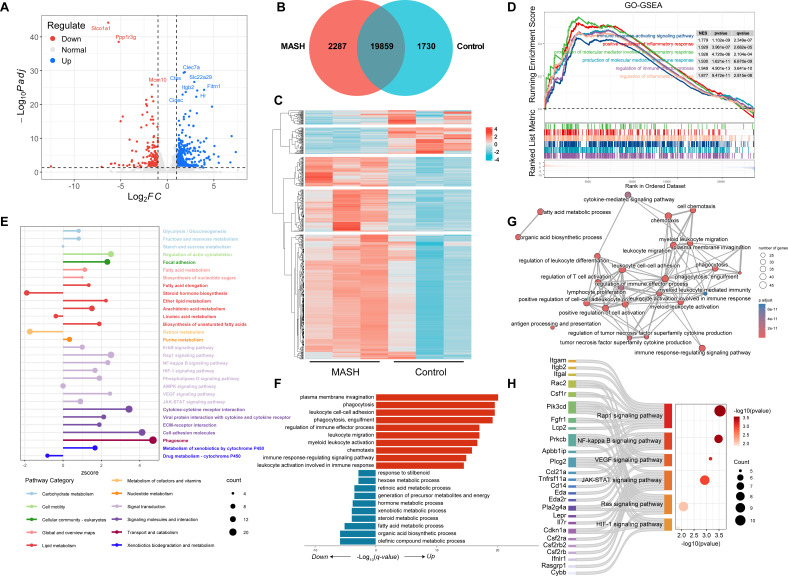
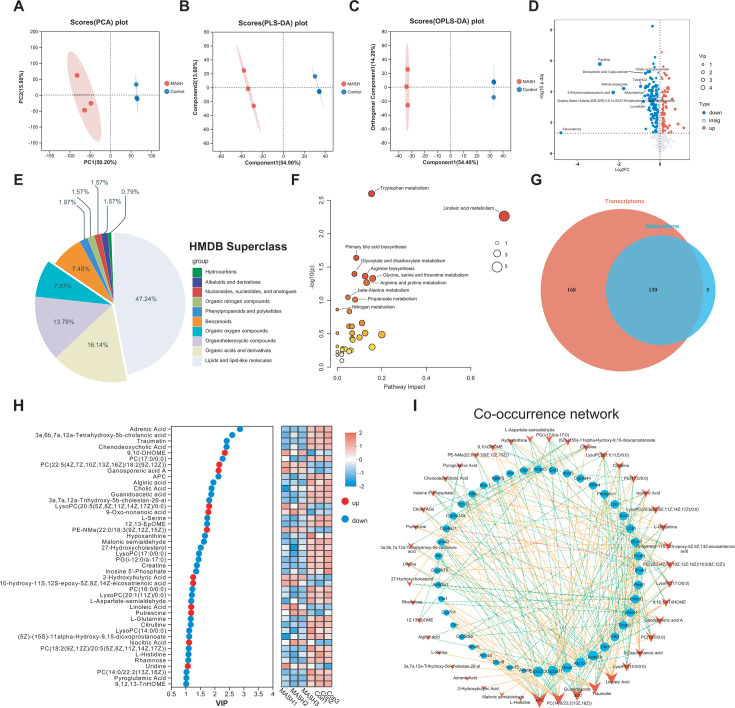
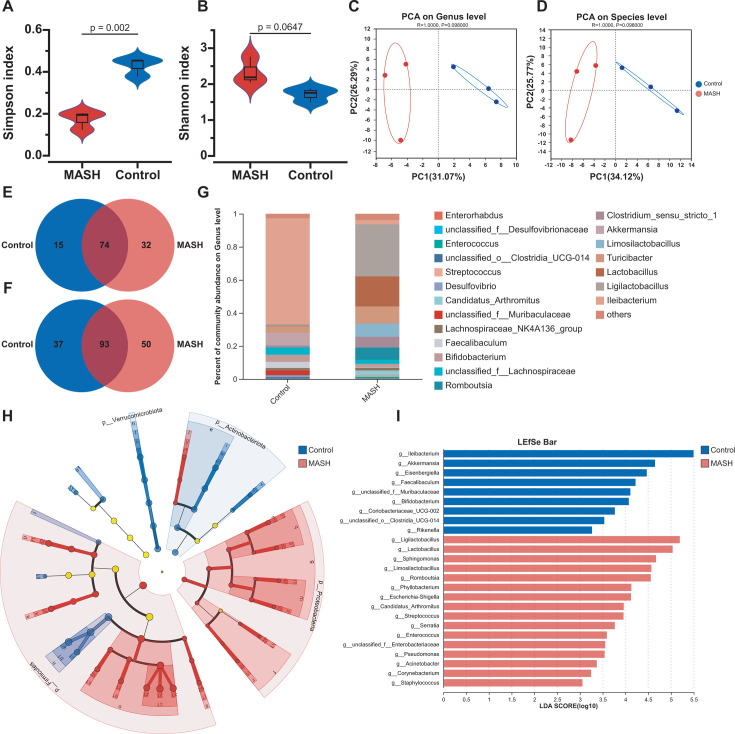
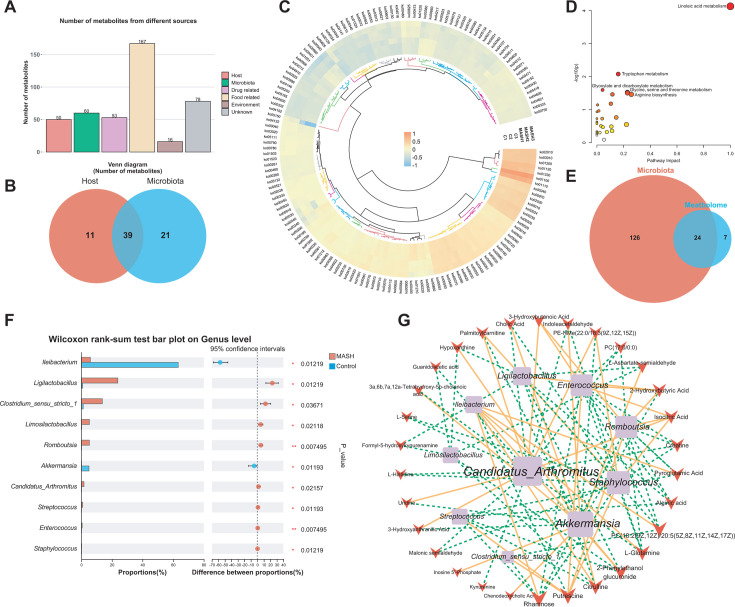
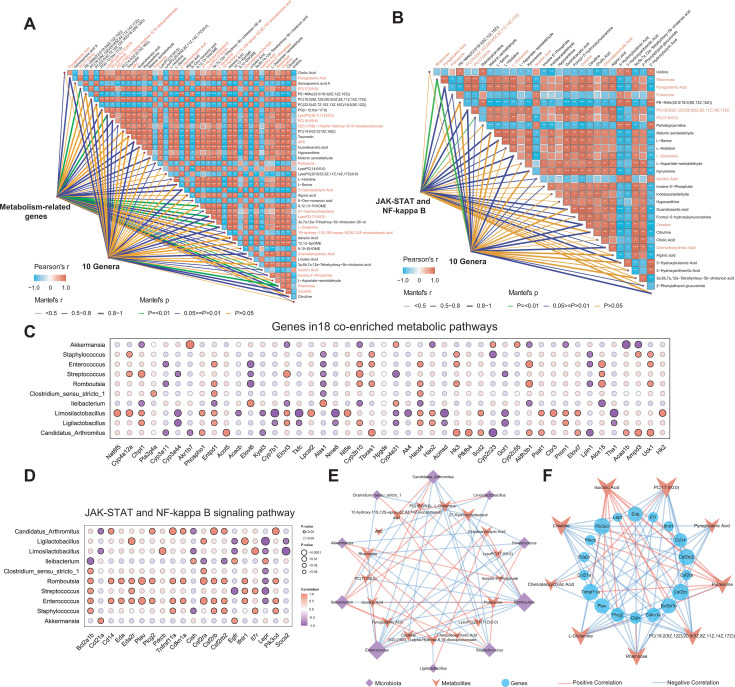
As a commonly known aggressive liver-related manifestation within the spectrum of metabolic syndrome with a significant risk of progressing to cirrhosis and hepatocellular carcinoma, metabolic dysfunction-associated steatohepatitis (MASH) is closely intertwined with obesity, insulin resistance, and dyslipidemia. Although the gut microbiota is implicated in MASH progression, the underlying mechanisms require further investigation. In this study, we sought to combine the analysis of the liver transcriptome, circulating metabolome, and gut microbiota to investigate the potential molecular mechanisms underlying the reciprocal regulation between gut microbiota and liver immune signaling. We utilized a high-fat and methionine/choline-deficient diet (HFMCD)-induced MASH model in a db/db mouse. Following annotation analysis using KEGG and Metorigin, a comprehensive correlation analysis was conducted among these genes and specific metabolites (such as L-glutamine, isocitric acid, putrescine, pyroglutamic acid, rhamnose) and gut microbiota genera (Enteroccus and Romboutsia). The results revealed intricate interactions among the liver's immune microenvironment, the metabolome, and the gut microbiota. These interactions suggest a potential regulatory mechanism for metabolic disorders and immune responses.IMPORTANCEOur multi-omics analysis showed that the interactions between gut microbiota and liver immune responses mediated by the disorders in lipid, amino acid, and glucose metabolism are associated with activation of the JAK-STAT and NF-κB signaling pathway in MASH. The multi-omics analysis provides valuable insights into the interactions among microbiota, circulating metabolites, and immune signaling. These insights can be harnessed to enhance the management of MASH.
Keywords: MASH; gut microbiota; metabolome; multi-omics analysis; transcriptome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
HBOT alleviates diet-induced MASH by reprograming gut microbiota and liver metabolism in mice.Free Radic Biol Med. 2025 Sep;237:600-614. doi: 10.1016/j.freeradbiomed.2025.05.420. Epub 2025 May 31. Free Radic Biol Med. 2025. PMID: 40456496
-
Alisol B ameliorated metabolic dysfunction-associated steatotic liver disease via regulating purine metabolism and restoring the gut microbiota disorders.Phytomedicine. 2025 Sep;145:156992. doi: 10.1016/j.phymed.2025.156992. Epub 2025 Jun 22. Phytomedicine. 2025. PMID: 40582207
-
Multi-omics analysis reveals associations between host gene expression, gut microbiota, and metabolites in chickens.J Anim Sci. 2024 Jan 3;102:skae263. doi: 10.1093/jas/skae263. J Anim Sci. 2024. PMID: 39243135
-
Management of urinary stones by experts in stone disease (ESD 2025).Arch Ital Urol Androl. 2025 Jun 30;97(2):14085. doi: 10.4081/aiua.2025.14085. Epub 2025 Jun 30. Arch Ital Urol Androl. 2025. PMID: 40583613 Review.
-
Gut microbiota and metabolomics in metabolic dysfunction-associated fatty liver disease: interaction, mechanism, and therapeutic value.Front Cell Infect Microbiol. 2025 Jul 23;15:1635638. doi: 10.3389/fcimb.2025.1635638. eCollection 2025. Front Cell Infect Microbiol. 2025. PMID: 40771314 Free PMC article. Review.
References
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
