

Patterns of intra- and intertumor phenotypic heterogeneity in lethal prostate cancer
- PMID: 40493417
- PMCID: PMC12321404
- DOI: 10.1172/JCI186599
Patterns of intra- and intertumor phenotypic heterogeneity in lethal prostate cancer
Abstract
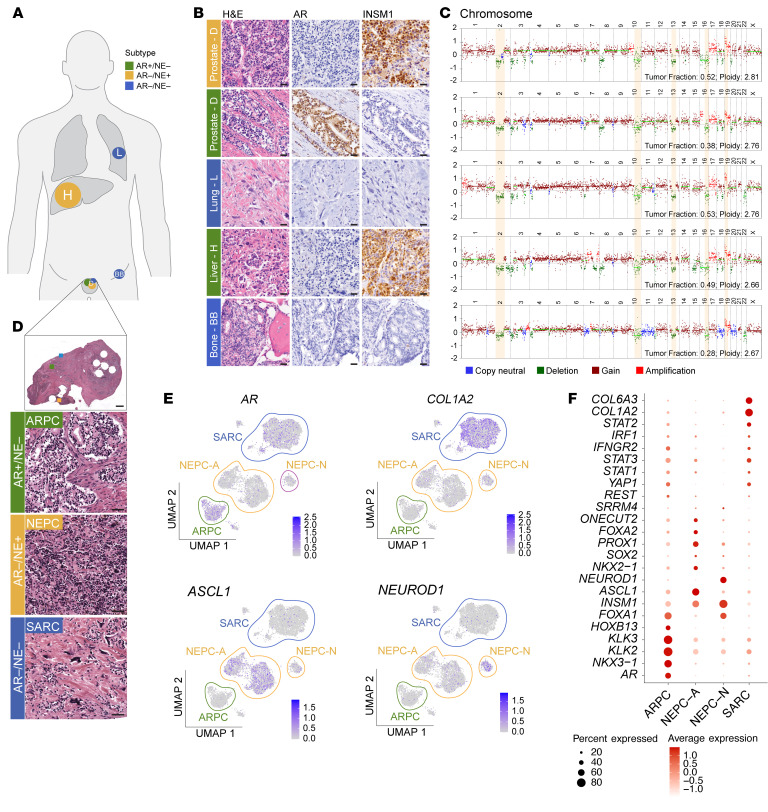
Metastatic prostate cancer (mPC) is a clinically and molecularly heterogeneous disease. While there is increasing recognition of diverse tumor phenotypes across patients, less is known about the molecular and phenotypic heterogeneity present within an individual. In this study, we aimed to define the patterns, extent, and consequences of inter- and intratumoral heterogeneity in lethal prostate cancer. By combining and integrating in situ tissue-based and sequencing approaches, we analyzed over 630 tumor samples from 52 patients with mPC. Our efforts revealed phenotypic heterogeneity at the patient, metastasis, and cellular levels. We observed that intrapatient intertumoral molecular subtype heterogeneity was common in mPC and showed associations with genomic and clinical features. Additionally, cellular proliferation rates varied within a given patient across molecular subtypes and anatomic sites. Single-cell sequencing studies revealed features of morphologically and molecularly divergent tumor cell populations within a single metastatic site. These data provide a deeper insight into the complex patterns of tumoral heterogeneity in mPC with implications for clinical management and the future development of diagnostic and therapeutic approaches.
Keywords: Cell biology; Molecular pathology; Oncology; Prostate cancer; Urology.
Conflict of interest statement
Figures





Comment in
- Uncovering phenotypic heterogeneity via research autopsy in lethal prostate cancer
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
