

Aberrant Tryptophan Metabolism Manipulates Osteochondral Homeostasis
- PMID: 40496774
- PMCID: PMC12150400
- DOI: 10.34133/research.0728
Aberrant Tryptophan Metabolism Manipulates Osteochondral Homeostasis
Abstract
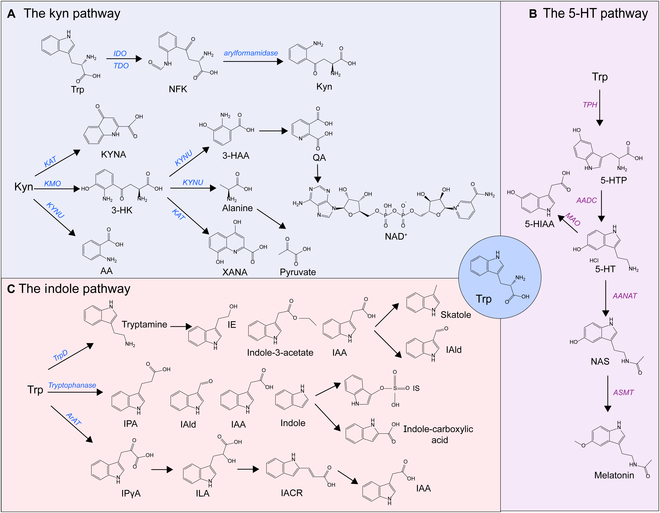
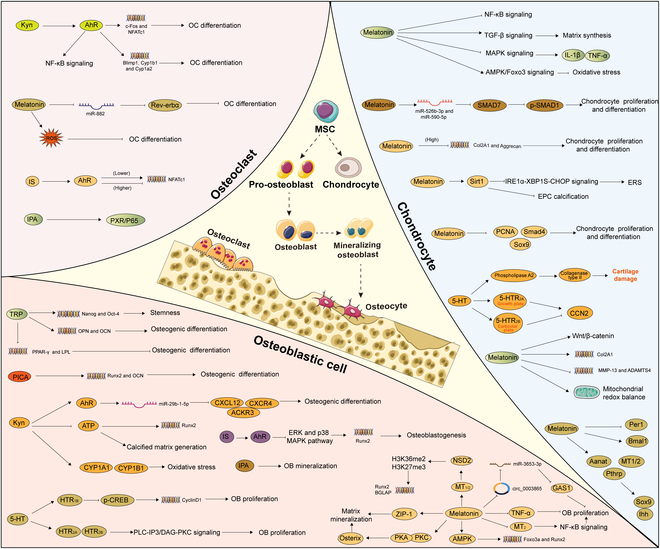
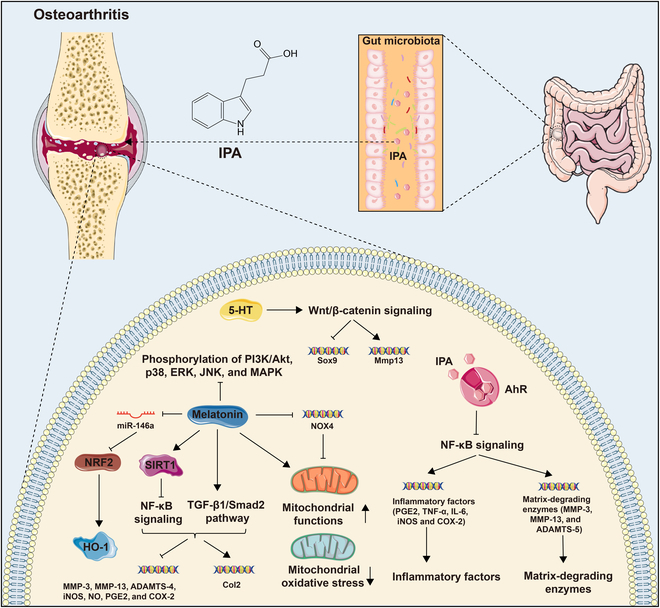
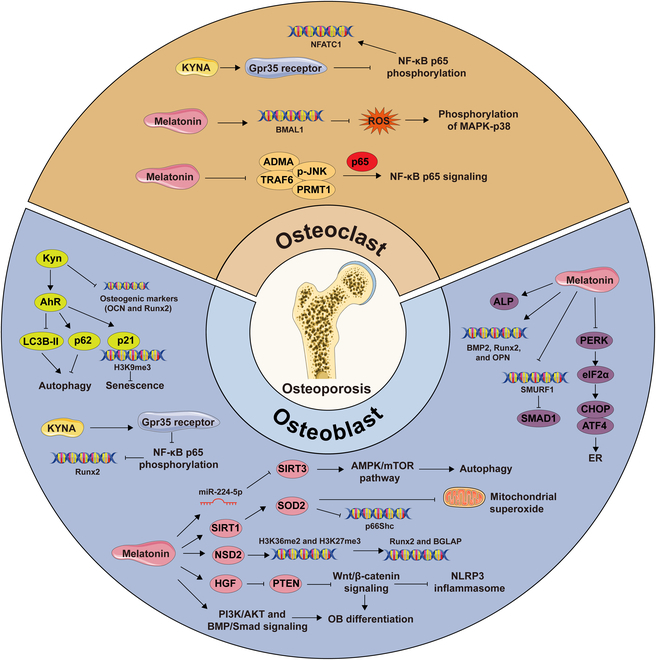
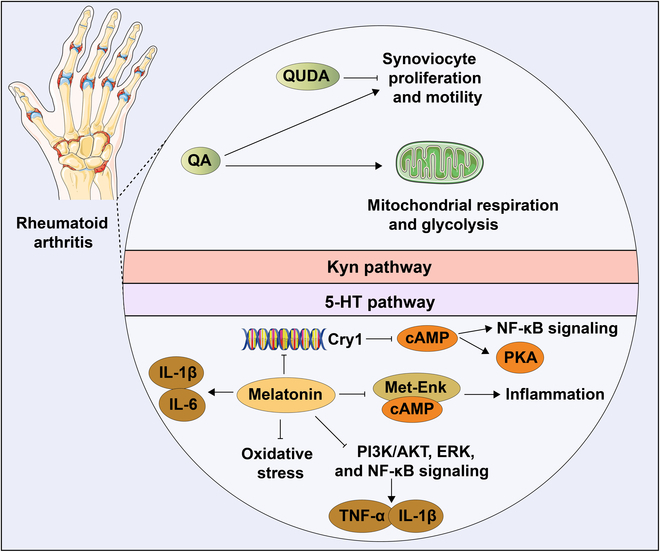
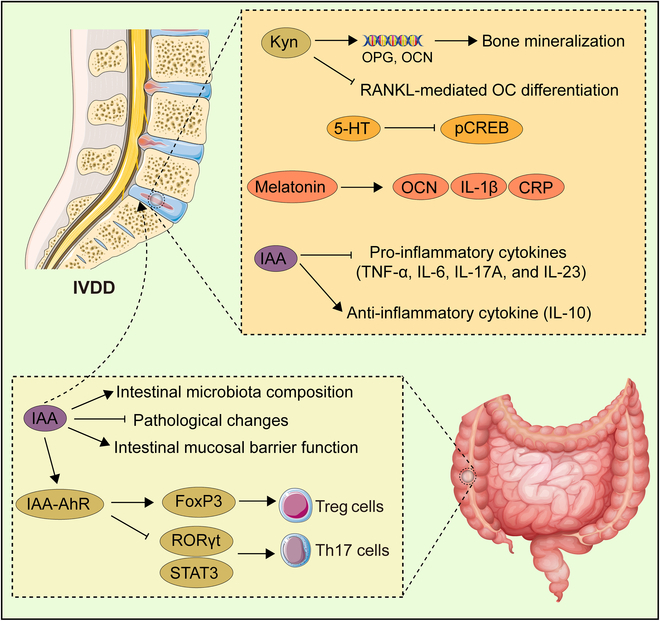
Tryptophan (Trp), an essential amino acid, performs as a precursor for synthesizing various bioactive molecules primarily metabolized through the kynurenine (Kyn), serotonin, and indole pathways. The diverse metabolites were deeply implicated in multiple physiological processes. Emerging research has revealed the multifaceted contribution of Trp in skeletal health and pathophysiology of bone-related disease with the involvement of specific receptors including aryl hydrocarbon receptor (AhR), which modulated the downstream signaling pathways to manage the expression of pivotal genes and thereby altered cellular biological processes, such as proliferation and differentiation. Accompanied by distinct alterations in immune function, inflammatory responses, endocrine balance, and other physiological aspects, their impact and efficacy in osteochondrogenic disorders have also been well documented. Nevertheless, a thorough understanding of Trp metabolism within bone biology is currently lacking. In this review, we elucidate the complexities of Trp metabolic pathway and several metabolites, delineating their versatile modulatory roles in the physiology and pathology of osteoblasts (OBs), osteoclasts (OCs), chondrocytes, and intercellular coupling effects, as well as in the progression of osteochondral disorder. Moreover, we comprehensively delineate the regulatory mechanisms by which gut microbiota-generated indole derivatives mediate bidirectional crosstalk along the gut-bone axis. The establishment of an elaborate governing network about bone homeostasis provides a novel insight on therapeutic interventions.
Copyright © 2025 Tingwen Xiang et al.
Conflict of interest statement
Competing interests: The authors declare that they have no competing interests.
Figures
References
-
- Wu G. Amino acids: Metabolism, functions, and nutrition. Amino Acids. 2009;37(1):1–17. - PubMed
-
- Vécsei L, Szalárdy L, Fülöp F, Toldi J. Kynurenines in the CNS: Recent advances and new questions. Nat Rev Drug Discov. 2013;12(1):64–82. - PubMed
-
- Agus A, Planchais J, Sokol H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe. 2018;23(6):716–724. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
