

Endothelial CLEC5A drives barrier dysfunction and vascular leakage responsible for lung injury in bacterial pneumonia and sepsis
- PMID: 40498836
- PMCID: PMC12154197
- DOI: 10.1126/sciadv.adt7589
Endothelial CLEC5A drives barrier dysfunction and vascular leakage responsible for lung injury in bacterial pneumonia and sepsis
Abstract
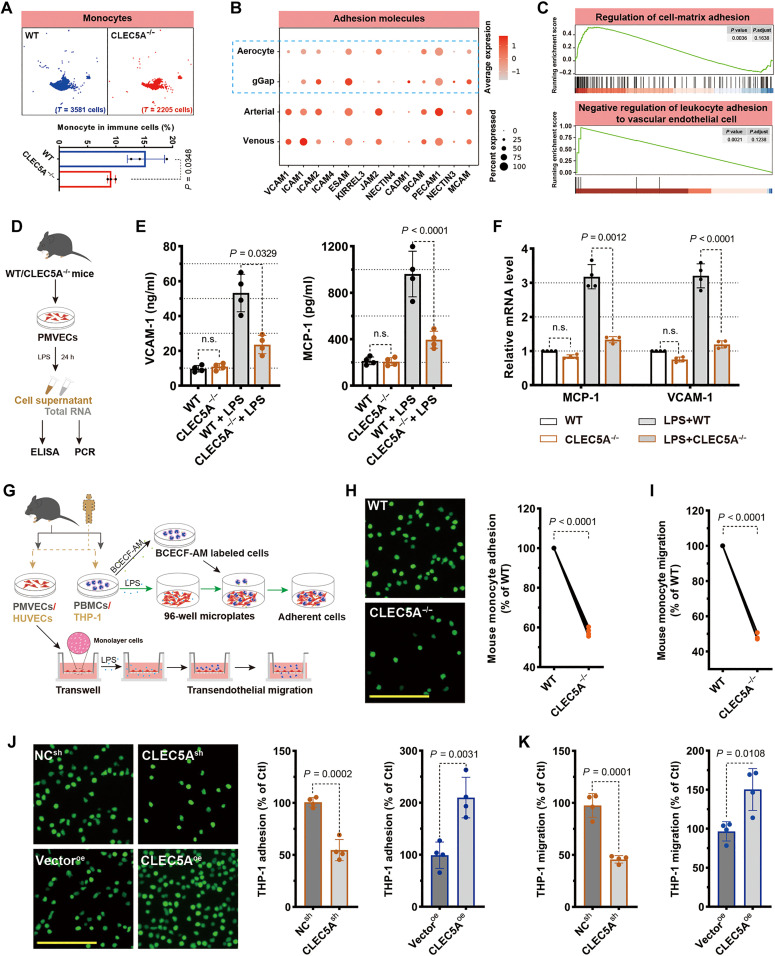
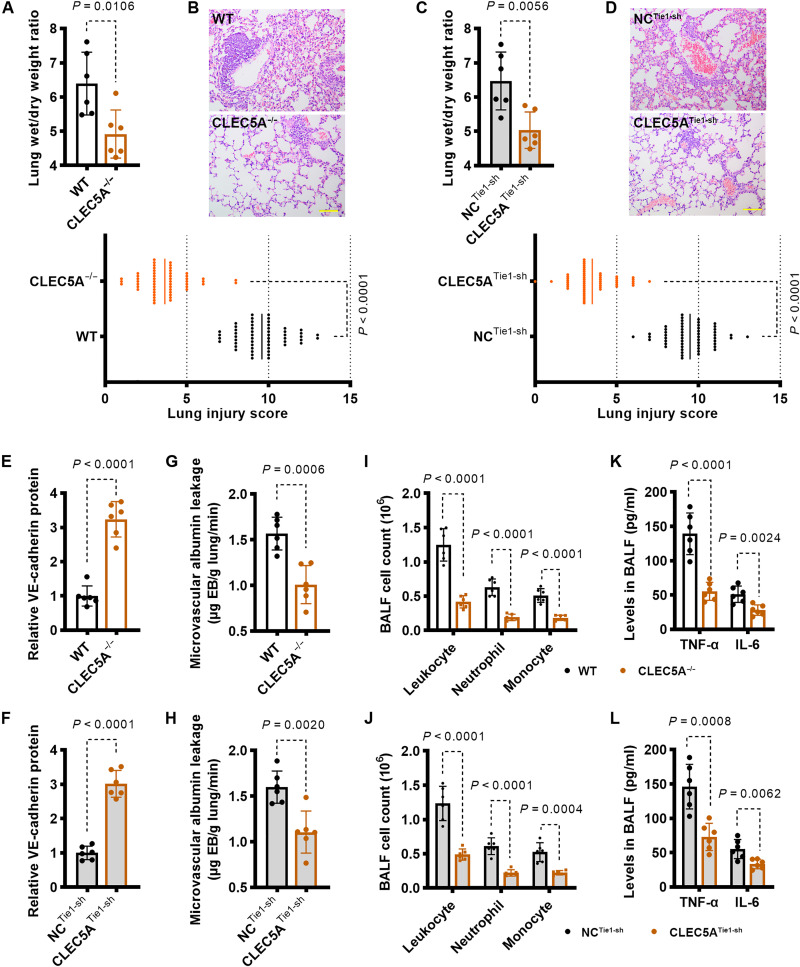
Endothelial barrier dysfunction and the resulting vascular injury are responsible for multiorgan failure in sepsis. Myeloid C-type lectin domain family 5 member A (CLEC5A) is a pattern recognition receptor involved in host defense against infection. Mice lacking CLEC5A were resistant to cecal ligation and puncture (CLP)-induced polymicrobial sepsis and lipopolysaccharide (LPS)-induced endotoxemia, as observed by decreased mortality. Single-cell RNA sequencing revealed transcriptomic heterogeneity of vascular endothelial cells in CLEC5A-deficient lungs following CLP. Endothelial-specific knockdown of CLEC5A improved survival of CLP-challenged mice, which was completely ineffective with reexpression of endothelial CLEC5A. The survival benefits were attributed to alleviated inflammatory storm and vascular leakage. Furthermore, endothelial CLEC5A deficiency protected mice against Escherichia coli-induced pneumonia. In vitro, CLEC5A deletion maintained trans-endothelial electrical resistance, and inhibited adhesion and trans-endothelial migration of monocytes/neutrophils under LPS stimulation. The study unveils the importance of CLEC5A in regulating endothelial barrier function and suggests endothelial CLEC5A as a therapeutic target for pneumonia or sepsis-causing bacterial infection.
Figures







References
-
- Singer M., Deutschman C. S., Seymour C. W., Shankar-Hari M., Annane D., Bauer M., Bellomo R., Bernard G. R., Chiche J.-D., Coopersmith C. M., Hotchkiss R. S., Levy M. M., Marshall J. C., Martin G. S., Opal S. M., Rubenfeld G. D., van der Poll T., Vincent J.-L., Angus D. C., The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315, 801–810 (2016). - PMC - PubMed
-
- Deutschman C. S., Tracey K. J., Sepsis: Current dogma and new perspectives. Immunity 40, 463–475 (2014). - PubMed
-
- Keeley A., Hine P., Nsutebu E., The recognition and management of sepsis and septic shock: A guide for non-intensivists. Postgrad. Med. J. 93, 626–634 (2017). - PubMed
-
- Cecconi M., Evans L., Levy M., Rhodes A., Sepsis and septic shock. Lancet 392, 75–87 (2018). - PubMed
-
- Sevransky J. E., Levy M. M., Marini J. J., Mechanical ventilation in sepsis-induced acute lung injury/acute respiratory distress syndrome: An evidence-based review. Crit. Care Med. 32, S548–S553 (2004). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
