

Epigenetic Heritability of Cell Plasticity Drives Cancer Drug Resistance through a One-to-Many Genotype-to-Phenotype Paradigm
- PMID: 40499006
- PMCID: PMC12314525
- DOI: 10.1158/0008-5472.CAN-25-0999
Epigenetic Heritability of Cell Plasticity Drives Cancer Drug Resistance through a One-to-Many Genotype-to-Phenotype Paradigm
Abstract
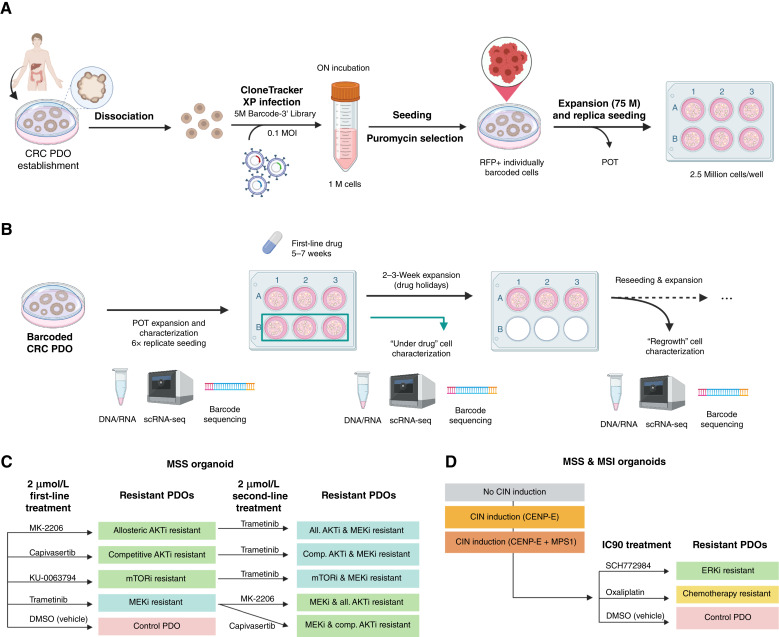
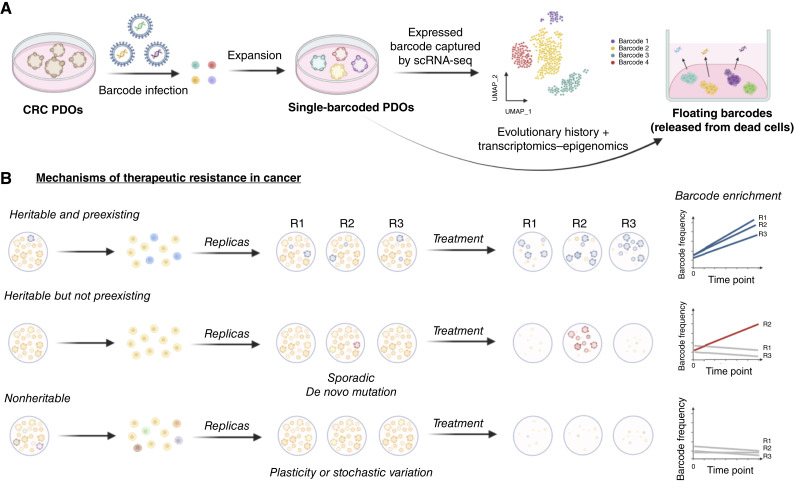
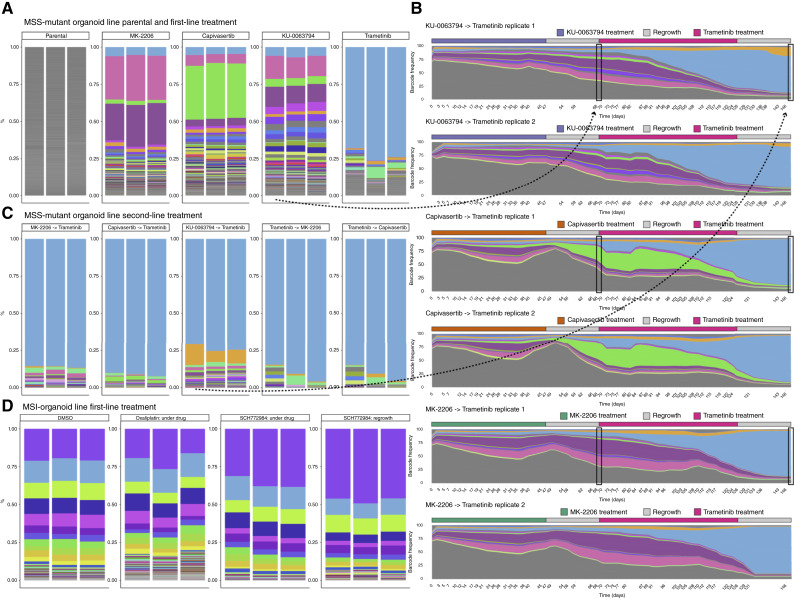
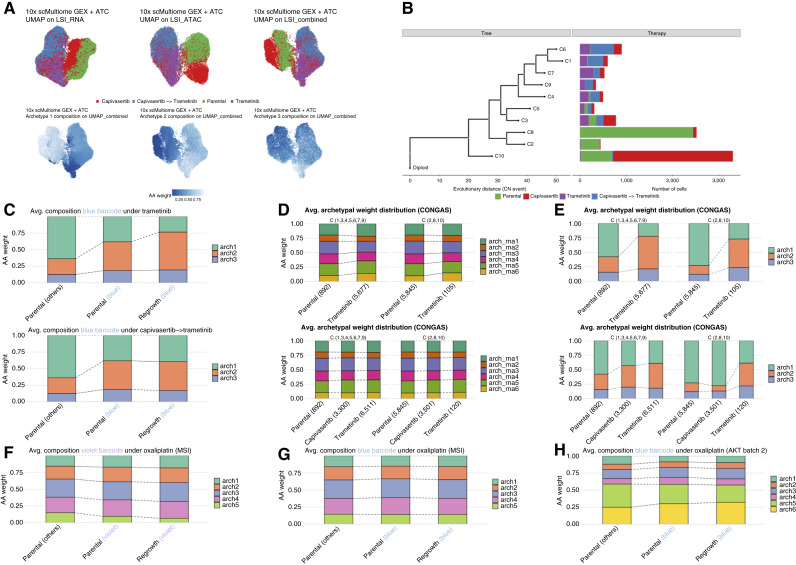
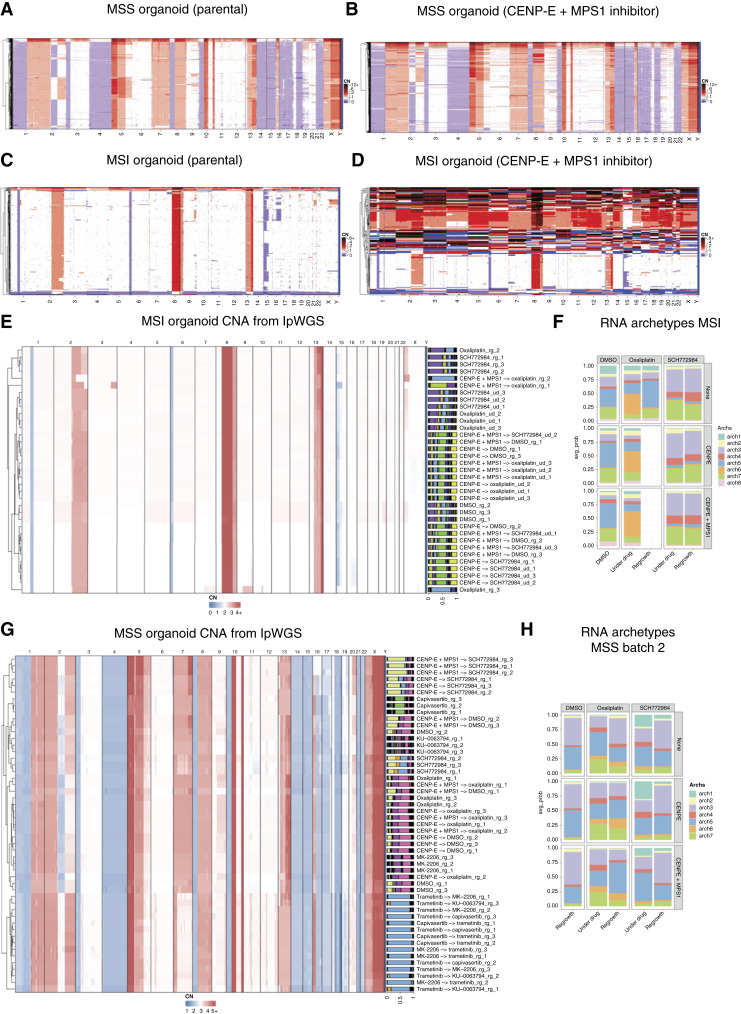
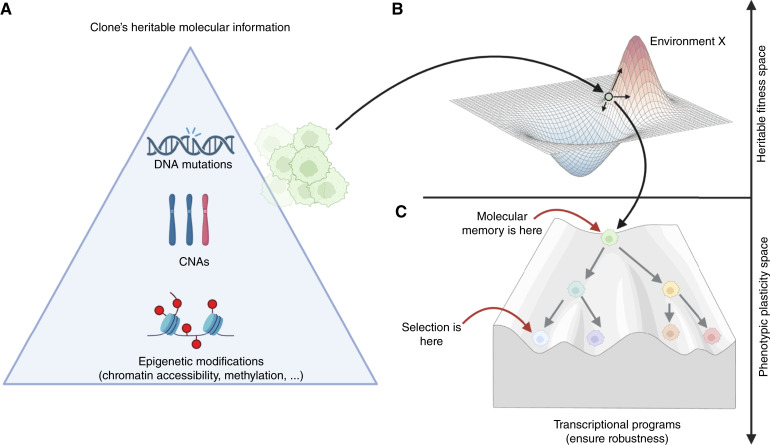
Cancer drug resistance is multifactorial, driven by heritable (epi)genetic changes but also by phenotypic plasticity. In this study, we dissected the drivers of resistance by perturbing organoids derived from patients with colorectal cancer longitudinally with drugs in sequence. Combined longitudinal lineage tracking, single-cell multiomics analysis, evolutionary modeling, and machine learning revealed that different targeted drugs select for distinct subclones, supporting rationally designed drug sequences. The cellular memory of drug resistance was encoded as a heritable epigenetic configuration from which multiple transcriptional programs could run, supporting a one-to-many (epi)genotype-to-phenotype map that explains how clonal expansions and plasticity manifest together. This epigenetic landscape may ensure drug-resistant subclones can exhibit distinct phenotypes in changing environments while still preserving the cellular memory encoding for their selective advantage. Chemotherapy resistance was instead entirely driven by transient phenotypic plasticity rather than stable clonal selection. Inducing further chromosomal instability before drug application changed clonal evolution but not convergent transcriptional programs. Collectively, these data show how genetic and epigenetic alterations are selected to engender a "permissive epigenome" that enables phenotypic plasticity.
Significance: Drug resistance is driven by genetic-epigenetic memory that enables cancer cells to adopt multiple phenotypic states depending on environmental conditions, supporting integration of evolutionary principles into biomarker discovery and personalized treatment strategies. This article is part of a special series: Driving Cancer Discoveries with Computational Research, Data Science, and Machine Learning/AI.
©2025 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
L. Patruno reports grants from Cancer Research UK–Associazione Italiana per la Ricerca contro il Cancro during the conduct of the study. A. Bertotti reports grants from Associazione Italiana per la Ricerca contro il Cancro—Italian Association for Cancer Research—during the conduct of the study. T.A. Graham reports other support from Genentech and personal fees from DAiNA Therapeutics outside the submitted work and has patents for GB2305655.9, GB2317139.0, and GB2501439.0 pending. No disclosures were reported by the other authors.
Figures

References
MeSH terms
Grants and funding
- A19771/Cancer Research UK (CRUK)
- A18052/Cancer Research UK (CRUK)
- 28961/Fondazione AIRC per la ricerca sul cancro ETS (AIRC)
- 22790/Fondazione AIRC per la ricerca sul cancro ETS (AIRC)
- 21091/Fondazione AIRC per la ricerca sul cancro ETS (AIRC)
- A26815/Cancer Research UK (CRUK)
- CIG 334261/NIHR Biomedical Research Centre, Royal Marsden NHS Foundation Trust/Institute of Cancer Research (BRC)
- 24913/Fondazione AIRC per la ricerca sul cancro ETS (AIRC)
- 202778/Z/16/Z/Wellcome Trust (WT)
- 202778/B/16/Z/Wellcome Trust (WT)
- A22909/Cancer Research UK (CRUK)
- DRCNPG-May21_100001/Cancer Research UK (CRUK)
- Katherine and Douglas Longden Chair in Oncology/Department of Aeronautics, Imperial College London
- WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
