

New groups of highly divergent proteins in families as old as cellular life with important biological functions in the ocean
- PMID: 40500757
- PMCID: PMC12153180
- DOI: 10.1186/s40793-025-00697-3
New groups of highly divergent proteins in families as old as cellular life with important biological functions in the ocean
Abstract
Background: Metagenomics has considerably broadened our knowledge of microbial diversity, unravelling fascinating adaptations and characterising multiple novel major taxonomic groups, e.g. CPR bacteria, DPANN and Asgard archaea, and novel viruses. Such findings profoundly reshaped the structure of the known Tree of Life and emphasised the central role of investigating uncultured organisms. However, despite significant progresses, a large portion of proteins predicted from metagenomes remain today unannotated, both taxonomically and functionally, across many biomes and in particular in oceanic waters.
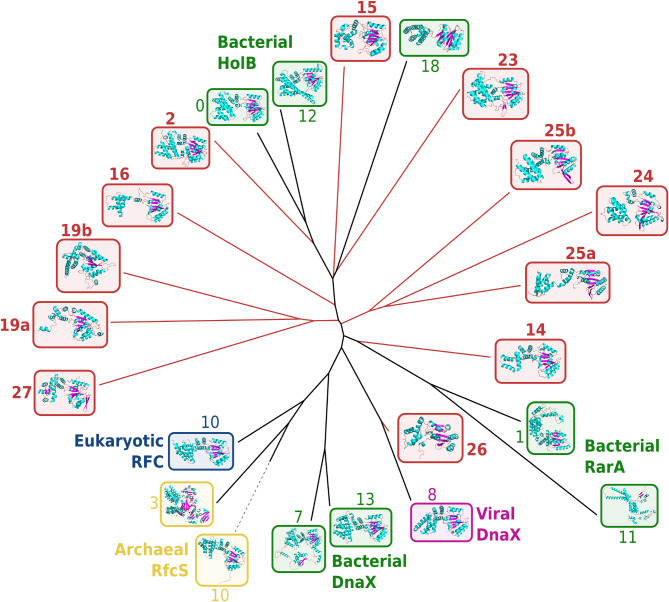
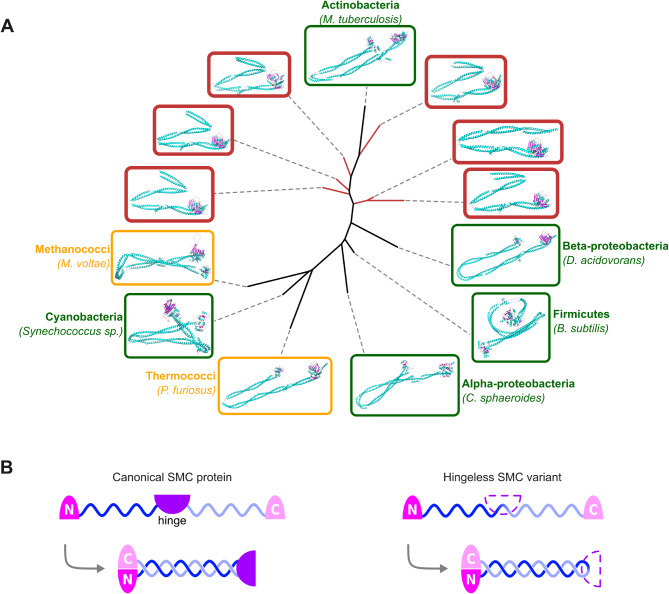
Results: Here, we used an iterative, network-based approach for remote homology detection, to probe a dataset of 40 million ORFs predicted in marine environments. We assessed the environmental diversity of 53 core gene families broadly distributed across the Tree of Life, with essential functions including translational, replication and trafficking processes. For nearly half of them, we identified clusters of remote environmental homologues that showed divergence from the known genetic diversity comparable to the divergence between Archaea and Bacteria, with representatives distributed across all the oceans. In particular, we report the detection of environmental clades with new structural variants of essential SMC (Structural Maintenance of Chromosomes) genes, divergent polymerase subunits forming deep-branching clades in the polymerase tree, and variant DNA recombinases in Bacteria as well as viruses.
Conclusions: These results indicate that significant environmental diversity may yet be unravelled even in strongly conserved gene families. Protein sequence similarity network approaches, in particular, appear well-suited to highlight potential sources of biological novelty and make better sense of microbial dark matter across taxonomical scales.
Keywords: Distant homology; Microbial dark matter; Microbiome; Sequence similarity networks.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures





References
-
- Staley JT, Konopka A. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol. 1985;39:321–46. - PubMed
-
- Alain K, Querellou J. Cultivating the uncultured: limits, advances and future challenges. Extremophiles. 2009;13:583–94. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources