

EvANI benchmarking workflow for evolutionary distance estimation
- PMID: 40501070
- PMCID: PMC12159288
- DOI: 10.1093/bib/bbaf267
EvANI benchmarking workflow for evolutionary distance estimation
Abstract
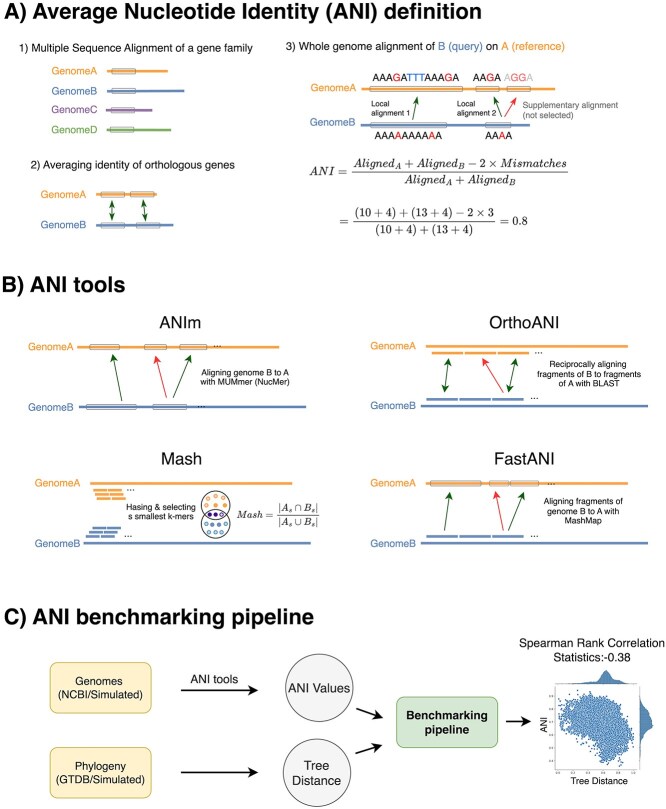
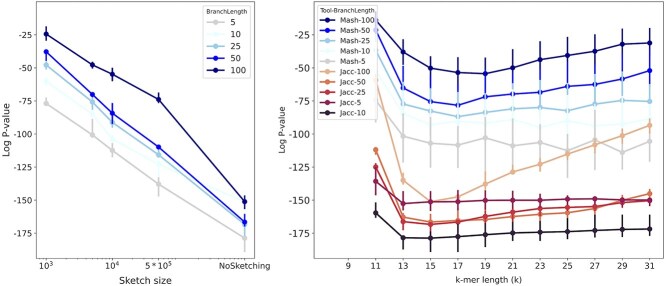
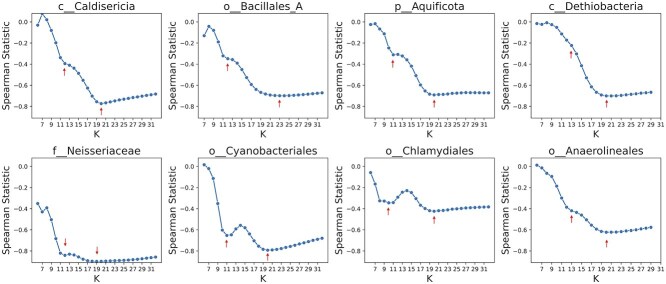
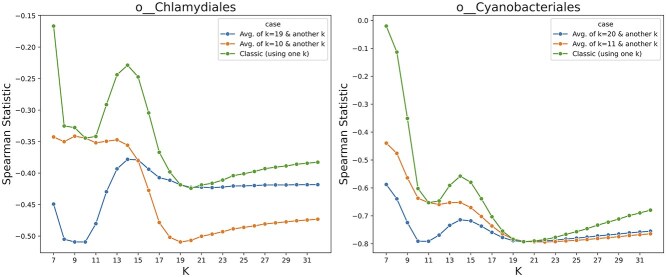
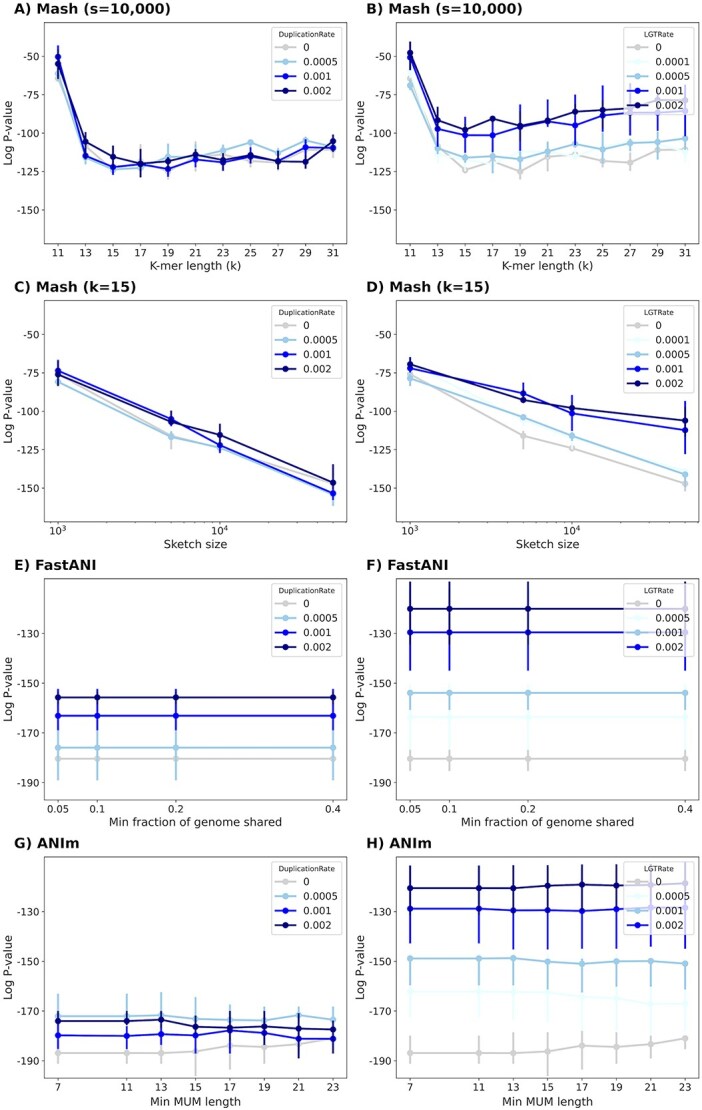
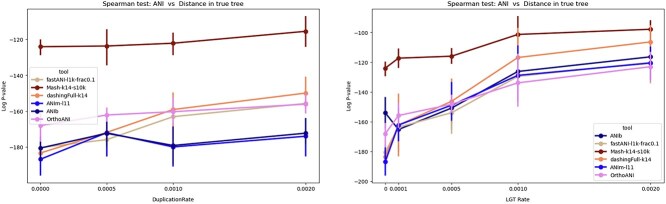
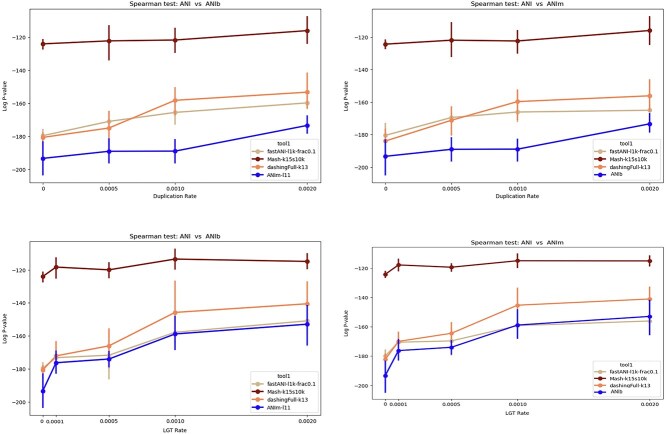
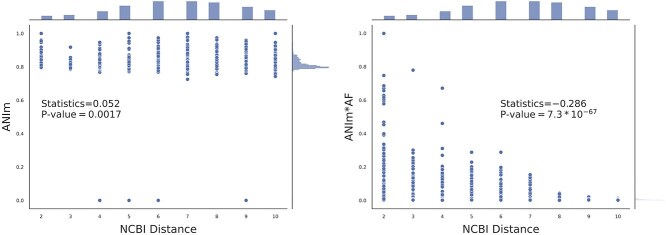
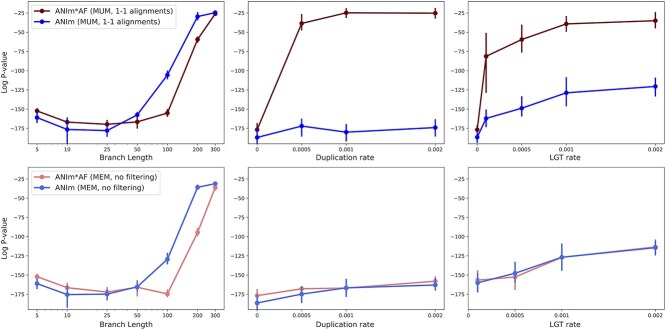
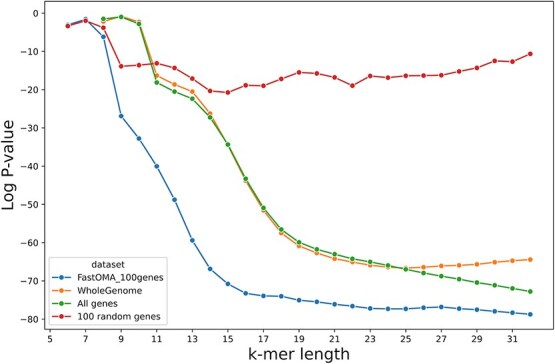
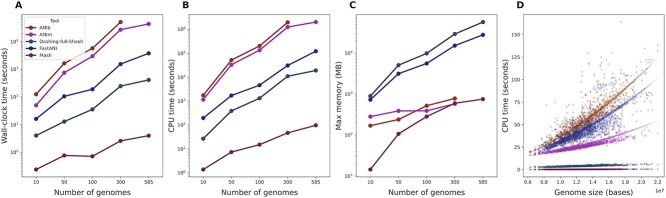
Advances in long-read sequencing technology have led to a rapid increase in high-quality genome assemblies. These make it possible to compare genome sequences across the Tree of Life, deepening our understanding of evolutionary relationships. Average nucleotide identity (ANI) is a metric for estimating the genetic similarity between two genomes, usually calculated as the mean identity of their shared genomic regions. These regions are typically found with genome aligners like Basic Local Alignment Search Tool BLAST or MUMmer. ANI has been applied to species delineation, building guide trees, and searching large sequence databases. Since computing ANI via genome alignment is computationally expensive, the field has increasingly turned to sketch-based approaches that use assumptions and heuristics to speed this up. We propose a suite of simulated and real benchmark datasets, together with a rank-correlation-based metric, to study how these assumptions and heuristics impact distance estimates. We call this evaluation framework EvANI. With EvANI, we show that ANIb is the ANI estimation algorithm that best captures tree distance, though it is also the least efficient. We show that k-mer-based approaches are extremely efficient and have consistently strong accuracy. We also show that some clades have inter-sequence distances that are best computed using multiple values of $k$, e.g. $k=10$ and $k=19$ for Chlamydiales. Finally, we highlight that approaches based on maximal exact matches may represent an advantageous compromise, achieving an intermediate level of computational efficiency while avoiding over-reliance on a single fixed k-mer length.
Keywords: BLAST; average nucleotide identity; evolution; genome; k-mer; sketching.
© The Author(s) 2025. Published by Oxford University Press.
Figures
Update of
-
EvANI benchmarking workflow for evolutionary distance estimation.bioRxiv [Preprint]. 2025 Feb 23:2025.02.23.639716. doi: 10.1101/2025.02.23.639716. bioRxiv. 2025. Update in: Brief Bioinform. 2025 May 1;26(3):bbaf267. doi: 10.1093/bib/bbaf267. PMID: 40027788 Free PMC article. Updated. Preprint.
References
-
- Hunt M, Lima L, Shen W. et al. Allthebacteria-all bacterial genomes assembled, available and searchable. Preprint bioRxiv. 2024;2024–03. 10.1101/2024.03.08.584059 - DOI
-
- Jukes TH, Cantor CR (1969) Evolution of protein molecules. In: Munro HN (ed) Mammalian Protein Metabolism, 21–132. 10.1016/B978-1-4832-3211-9.50009-7 - DOI
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
